Retavase Prescribing Information
Package insert / product label
Generic name: reteplase, recombinant
Dosage form: injection
Drug class: Thrombolytics
J Code (medical billing code): J2993 (18.8 mg, intravenous)
Medically reviewed by Drugs.com. Last updated on Apr 24, 2022.
On This Page
- Indications and Usage
- Dosage and Administration
- Dosage Forms and Strengths
- Contraindications
- Warnings and Precautions
- Adverse Reactions/Side Effects
- Use In Specific Populations
- Description
- Clinical Pharmacology
- Nonclinical Toxicology
- Clinical Studies
- How Supplied/Storage and Handling
Highlights of Prescribing Information
These highlights do not include all the information needed to use RETAVASE safely and effectively. See full prescribing information for RETAVASE.
RETAVASE (reteplase) for injection, for intravenous use
Initial U.S. Approval: 1996
Indications and Usage for Retavase
RETAVASE is a tissue plasminogen activator (tPA) indicated for treatment of acute ST-elevation myocardial infarction (STEMI) to reduce the risk of death and heart failure. (1)
Limitation of Use: The risk of stroke may outweigh the benefit produced by thrombolytic therapy in patients whose STEMI puts them at low risk for death or heart failure. (1)
Retavase Dosage and Administration
- Two 10 unit intravenous injections, each administered over 2 minutes, 30 minutes apart. (2.1)
- No other medication should be injected or infused simultaneously via the same intravenous line or added to the injection solution. (2.1)
Dosage Forms and Strengths
For Injection: 10 units as a lyophilized powder in single-use vials for reconstitution co-packaged with Sterile Water for Injection, USP in 10 mL prefilled syringe. (3)
Contraindications
-
Do not use in patients with:
- Active internal bleeding (4)
- Recent stroke (4)
- Recent intracranial or intraspinal surgery or serious head trauma (4)
- Intracranial neoplasm, arteriovenous malformation, or aneurysm (4)
- Known bleeding diathesis (4)
- Severe uncontrolled hypertension (4)
Warnings and Precautions
- Increases the risk of bleeding. Avoid intramuscular injections. (5.1)
- Hypersensitivity (5.2)
- Cholesterol embolism (5.3)
Adverse Reactions/Side Effects
The most common adverse reaction (>5%) is bleeding. (6.1)
To report SUSPECTED ADVERSE REACTIONS, contact Chiesi USA, Inc. at 1-888-661-9260 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
Use In Specific Populations
- Pediatric Use: Safety and effectiveness have not been established. (8.4)
Revised: 4/2022
Full Prescribing Information
1. Indications and Usage for Retavase
RETAVASE is indicated for use in acute ST-elevation myocardial infarction (STEMI) to reduce the risk of death and heart failure.
Limitation of Use: The risk of stroke may outweigh the benefit produced by thrombolytic therapy in patients whose STEMI puts them at low risk for death or heart failure.
2. Retavase Dosage and Administration
2.1 Dosing Information and Administration
As soon as possible after the onset of STEMI, administer 10 units intravenously over 2 minutes. Administer a second dose of 10 units 30 minutes after the first dose.
2.2 Reconstitution
Reconstitute RETAVASE immediately before administration.
Reconstitute RETAVASE only with the supplied Sterile Water for Injection. Slight foaming upon reconstitution may occur; if necessary allow the vial to stand undisturbed for several minutes to allow dissipation of any large bubbles. Prior to administration, inspect the product for particulate matter and discoloration.
Use aseptic technique throughout.
Step 1: Open the package containing the reconstitution spike. Remove the protective cap from the luer lock port of the reconstitution spike and remove the protective cap on the end of the sterile 10 mL pre-filled syringe. Remove the protective flip-cap from one vial of RETAVASE.
Step 2: Clean the rubber closure with an alcohol wipe (not contained within kit).
Step 3: Connect the sterile pre-filled syringe to the reconstitution spike.
Step 4: Remove the protective cap from the spike end of the reconstitution spike and firmly insert the spike into the vial of RETAVASE.
Step 5: Connect the syringe plunger to the sterile 10 mL pre-filled syringe by screwing the plunger into the rubber stopper.
Step 6: Transfer the 10 mL of Sterile Water for Injection through the reconstitution spike into the vial of RETAVASE.
Step 7: With the reconstitution spike and empty pre-filled syringe still attached to the vial, swirl the vial gently until the contents are fully dissolved, may take up to 2 minutes. DO NOT SHAKE. Upon reconstitution, the solution should be clear and colorless. Discard if particulate matter or discoloration is observed. The resulting solution concentration is 1 unit/mL and delivers 10 mL (10 units reteplase).
Step 8: Disconnect the empty pre-filled syringe from the reconstitution spike and connect the plastic, graduated syringe to the reconstitution spike that is still attached to the vial.
Step 9: Withdraw 10 mL of RETAVASE reconstituted solution into the graduated syringe. A small amount of solution will remain in the vial due to overfill. Detach the graduated syringe from the reconstitution spike.
2.3 Heparin Incompatibility
Heparin and RETAVASE are incompatible. Do not administer RETAVASE through an intravenous line containing heparin.
3. Dosage Forms and Strengths
For Injection: 10 units as a lyophilized powder in single-use vials for reconstitution co-packaged with Sterile Water for Injection, USP in 10 mL prefilled syringe.
4. Contraindications
Because thrombolytic therapy increases the risk of bleeding, RETAVASE is contraindicated in the following situations:
- Active internal bleeding
- Recent stroke
- Intracranial or intraspinal surgery or serious head trauma within 3 months
- Intracranial conditions that increase the risk of bleeding (e.g. neoplasms, arteriovenous malformation, or aneurysms)
- Bleeding diathesis
- Current severe uncontrolled hypertension
5. Warnings and Precautions
5.1 Bleeding
RETAVASE can cause significant and sometimes fatal bleeding. Avoid intramuscular injections and other trauma to a patient administered RETAVASE. Minimize venipunctures. Avoid puncturing noncompressible veins, such as the internal jugular and subclavian. If an arterial puncture is necessary, use an upper extremity vessel that is accessible to manual compression, apply pressure for at least 30 minutes, and monitor the puncture site closely. As fibrin is lysed during RETAVASE therapy, bleeding from recent puncture sites or other recent trauma may occur.
Should serious bleeding (not controllable by local pressure) occur, terminate concomitant anticoagulant therapy. Withhold the second RETAVASE dose if serious bleeding occurs before it is administered.
5.2 Hypersensitivity Reactions
Hypersensitivity reactions have been reported with RETAVASE administration. Signs and symptoms observed included rash, pruritis, erythema, glossal (tongue) edema, hypotension and respiratory distress. If an anaphylactoid reaction occurs, withhold the second dose of RETAVASE and initiate appropriate therapy.
5.3 Cholesterol Embolization
Cholesterol embolism has been reported in patients treated with thrombolytic agents. Cholesterol embolism may present with livedo reticularis, “purple toe” syndrome, acute renal failure, gangrenous digits, hypertension, pancreatitis, myocardial infarction, cerebral infarction, spinal cord infarction, retinal artery occlusion, bowel infarction, and rhabdomyolysis and can be fatal. It is also associated with invasive vascular procedures (e.g., cardiac catheterization, angiography, vascular surgery) and/or anticoagulant therapy.
5.4 Drug/Laboratory Test Interactions
Coagulation tests and measures of fibrinolytic activity are unreliable during RETAVASE therapy unless specific precautions are taken to prevent in vitro artifacts. When present in blood at pharmacologic concentrations, RETAVASE remains active under in vitro conditions, which can result in degradation of fibrinogen in blood samples removed for analysis.
6. Adverse Reactions/Side Effects
The following adverse reactions are discussed in greater detail in the other sections of the label:
- Bleeding [see Contraindications (4) and Warnings and Precautions (5.1)]
- Hypersensitivity Reactions [see Warnings and Precautions (5.2)]
- Cholesterol Embolization [see Warnings and Precautions (5.3)]
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Bleeding
The most frequent adverse reaction associated with RETAVASE is bleeding.
- Intracranial hemorrhage [see Clinical Studies (14)]
In the INJECT clinical trial, the overall rate of in-hospital, intracranial hemorrhage was 0.8%. The risk for intracranial hemorrhage is increased in patients with advanced age (2.2% among patients >70 years old) or with elevated blood pressure (2.4% among patients with systolic blood pressure >160 mmHg).
- Other types of hemorrhage
The incidence of other types of bleeding events in clinical studies of RETAVASE varied depending upon the use of arterial catheterization or other invasive procedures and whether the study was performed in Europe or the USA. The overall incidence of any bleeding event in patients treated with RETAVASE in clinical studies (n = 3,805) was 21.1%. The rates for bleeding events, regardless of severity, for the 10 + 10 unit RETAVASE regimen from controlled clinical studies are summarized in Table 1.
Table 1: RETAVASE Hemorrhage Rates
|
Bleeding Site |
INJECT |
RAPID 1 and RAPID 2 |
|
|
Europe |
USA |
Europe |
|
|
|||
|
Injection Site* |
4.6% |
48.6% |
19.5% |
|
Gastrointestinal |
2.5% |
9.0% |
1.8% |
|
Genitourinary |
1.6% |
9.5% |
0.9% |
|
Anemia, site unknown |
2.6% |
1.4% |
0.9% |
In these studies the severity and sites of bleeding events were similar for RETAVASE and the comparison thrombolytic agents.
Allergic Reactions
Among the 2,965 patients receiving RETAVASE in the INJECT trial, serious allergic reactions were noted in 3 patients, with one patient experiencing dyspnea and hypotension.
Among the 9,938 patients that received RETAVASE in a postmarketing clinical study, 8 patients experienced allergic and/or anaphylactoid reactions. Signs and symptoms observed included rash, pruritis, erythema, glossal (tongue) edema, hypotension, and respiratory distress.
8. Use In Specific Populations
8.1 Pregnancy
Risk Summary
Limited published data with RETAVASE use in pregnant women are insufficient to inform a drug associated risk of adverse developmental outcomes. In animal reproduction studies, reteplase administered to pregnant rabbits resulted in hemorrhaging in the genital tract, leading to abortions in mid-gestation in doses 3 times the human dose; however, there was no evidence of fetal anomalies in rats at doses up to 15 times the human dose.
The estimated background risk of major birth defects and miscarriage for the indicated population(s) are unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively.
Clinical Considerations
Maternal Adverse Reactions
The most common complication of thrombolytic therapy is bleeding and pregnancy may increase this risk.
Data
Animal Data
Reteplase was administered to pregnant rabbits in doses 3 times the human dose (0.86 units/kg) resulting in hemorrhaging in the genital tract, leading to abortions in mid-gestation. In animal developmental studies in rats at Reteplase doses up to 15 times the human dose (4.31 units/kg), there was no evidence of fetal anomalies.
8.2 Lactation
Risk Summary
There are no data on the presence of reteplase in either human or animal milk, the effects on the breastfed infant, or the effects on milk production. RETEVASE has not been studied in nursing mothers.
8.4 Pediatric Use
Safety and effectiveness of RETAVASE in pediatric patients have not been established.
11. Retavase Description
Reteplase is a non-glycosylated deletion mutein of tissue plasminogen activator (tPA), containing the kringle 2 and the protease domains of human tPA. Reteplase contains 355 of the 527 amino acids of native tPA (amino acids 1-3 and 176-527). Reteplase is produced by recombinant DNA technology in E. coli. The protein is isolated as inactive inclusion bodies from E. coli, converted into its active form by an in vitro folding process and purified by chromatographic separation. The molecular weight of Reteplase is 39,571 daltons.
Potency is expressed in units (U) using a reference standard which is specific for RETAVASE and is not comparable with units used for other thrombolytic agents.
RETAVASE (reteplase) for Injection is a sterile, white, lyophilized powder for intravenous injection after reconstitution with Sterile Water for Injection, USP (without preservatives). Following reconstitution with 10 mL of Sterile Water for Injection, the resulting concentration is 1 unit/mL to allow for delivery of 10 mL (10 units reteplase). The pH is 6.0 ± 0.3. RETAVASE is supplied with overfill to ensure sufficient drug for administration of each 10 unit injection.
Each single-use vial delivers:
|
Reteplase |
10 units |
|
Dipotassium Hydrogen Phosphate |
131 mg |
|
Phosphoric Acid |
49.3 mg |
|
Polysorbate 80 |
5 mg |
|
Sucrose |
350 mg |
|
Tranexamic Acid |
8 mg |
12. Retavase — Clinical Pharmacology
12.1 Mechanism of Action
RETAVASE is a recombinant plasminogen activator which catalyzes the cleavage of endogenous plasminogen to generate plasmin. Plasmin in turn degrades the fibrin matrix of the thrombus, thereby exerting its thrombolytic action.
12.2 Pharmacodynamics
In a controlled trial, 36 of 56 patients treated for myocardial infarction had a decrease in fibrinogen levels to below 100 mg/dL by 2 hours following the administration of RETAVASE as two intravenous injections (10 + 10 unit) in which 10 units was followed 30 minutes later by a second dose of 10 units. The mean fibrinogen level returned to the baseline value by 48 hours.
12.3 Pharmacokinetics
Based on the measurement of thrombolytic activity, RETAVASE is cleared from plasma at a rate of 250-450 mL/min, with an effective half-life of 13-16 minutes. RETAVASE is cleared primarily by the liver and kidney.
13. Nonclinical Toxicology
13.1 Carcinogenesis and Mutagenesis
Long-term studies in animals have not been performed to evaluate the carcinogenic potential of RETAVASE. Studies to determine mutagenicity, chromosomal aberrations, gene mutations, and micronuclei induction were negative at all concentrations tested.
14. Clinical Studies
RETAVASE was evaluated in three controlled clinical studies comparing RETAVASE to other thrombolytic agents. In all three studies, patients were treated with aspirin (initial doses of 160 mg to 350 mg and subsequent doses of 75 mg to 350 mg) and heparin (a 5,000 unit IV bolus prior to the administration of RETAVASE or control, followed by a 1000 unit/hour continuous IV infusion for at least 24 hours).
The INJECT study compared RETAVASE to streptokinase on mortality rates at 35 days following acute ST-elevation myocardial infarction (STEMI). INJECT was a double-blind study in which 6,010 patients with no more than 12 hours of chest pain consistent with coronary ischemia and either ST segment elevation or bundle branch block on ECG were randomized 1:1 to RETAVASE (10 + 10 unit) or streptokinase (1.5 million units over 60 minutes). Patients with cerebrovascular or other bleeding risks or with systolic blood pressure >200 mm Hg or diastolic blood pressure >100 mm Hg were excluded from enrollment. The study was designed to determine whether the effect of RETAVASE on survival was noninferior to that of streptokinase by ruling out with 95% confidence that 35-day mortality among RETAVASE patients was no more than 1% higher than among streptokinase patients. The results of the primary endpoint (mortality at 35 days), 6-month mortality, and selected other in-hospital endpoints are shown in Table 2.
Table 2: INJECT Study: Selected Results
|
Endpoint |
RETAVASE |
Streptokinase |
RETAVASE-Streptokinase ∆ (95% CI) |
|
|||
|
35-day mortality* |
8.9% |
9.4% |
-0.5 (-2.0, 0.9) |
|
6-month mortality |
11.0% |
12.1% |
-1.1 (-2.7, 0.6) |
|
Cardiogenic shock |
4.6% |
5.8% |
-1.2 (-2.4, -0.1) |
|
Heart failure in-hospital |
24.8% |
28.1% |
-3.3 (-5.6, -1.1) |
|
Any stroke in-hospital |
1.4% |
1.1% |
0.3 (-0.3, 0.8) |
|
Intracranial hemorrhage in-hospital |
0.8% |
0.4% |
0.4 (0.0, 0.8) |
More patients treated with RETAVASE experienced hemorrhagic strokes than did patients treated with streptokinase. An exploratory analysis indicated that the incidence of intracranial hemorrhage was higher among older patients or those with elevated blood pressure.
The other two studies (RAPID 1 and RAPID 2) compared coronary artery patency of RETAVASE to two regimens of alteplase in patients with STEMI. In RAPID 1 patients within 6 hours of the onset of symptoms were randomized to open-label administration of one of three regimens of RETAVASE (doses of 10 + 10 unit, 15 unit, or 10 + 5 unit) or to alteplase (100 mg over 3 hours). In RAPID 2 patients within 12 hours of the onset of symptoms were randomized to open-label administration of either RETAVASE (10 + 10 unit) or alteplase (100 mg over 1.5 hours). The primary endpoint for both studies was patency of the infarct-related artery 90 minutes after initiation of therapy. Interpretation of coronary angiograms was blinded.
A higher percentage of subjects administered RETEVASE had complete flow (TIMI grade 3) and partial or complete flow (TIMI grades 2 or 3) compared to both regimens of alteplase. The relationship between coronary artery patency and clinical efficacy has not been established.
In both clinical trials the re-occlusion rates were similar for RETAVASE and alteplase.
Table 3: RAPID 1 and RAPID 2 Studies: Angiographic Results
|
90 minute patency rates |
RAPID 2 |
RAPID 1* |
||||
|
RETAVASE N = 157 |
Alteplase N = 146 |
p— value |
RETAVASE N = 142 |
Alteplase N = 145 |
p— value |
|
|
||||||
|
TIMI 2 or 3 |
83% |
73% |
0.03 |
85% |
77% |
0.08 |
|
TIMI 3 |
60% |
45% |
0.01 |
63% |
49% |
0.02 |
16. How is Retavase supplied
RETAVASE (reteplase) for Injection is supplied as a sterile, preservative-free, lyophilized powder in 10 unit vials without a vacuum, in the following packaging configurations:
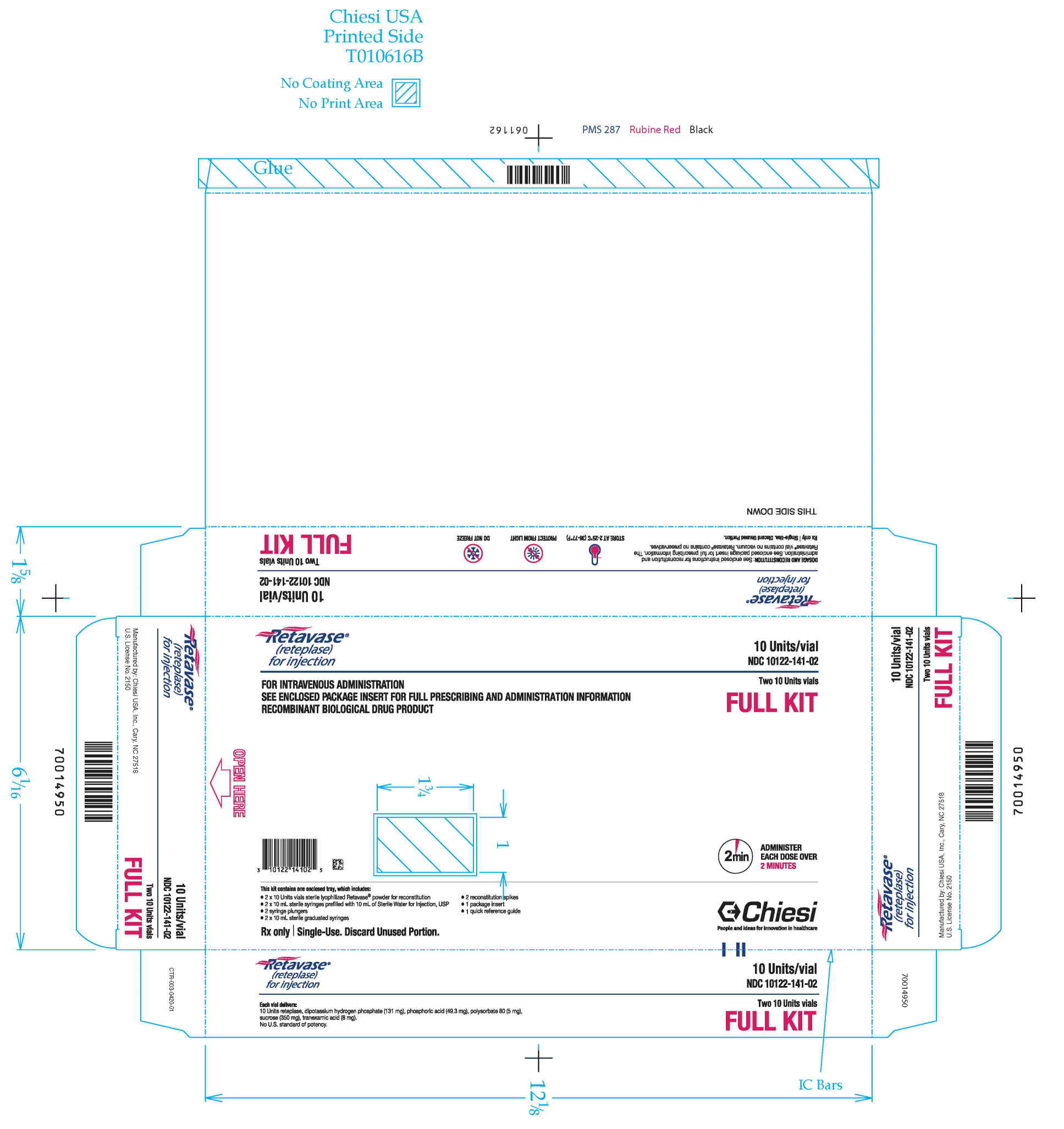
RETAVASE Kit (NDC 10122-141-02): 2 single-use RETAVASE vials 10 units, 2 single-use prefilled syringes for reconstitution (10 mL Sterile Water for Injection, USP), 2 syringe plungers, 2 sterile 10 mL graduated syringes, 2 sterile reconstitution spikes, 1 quick reference guide and 1 package insert.
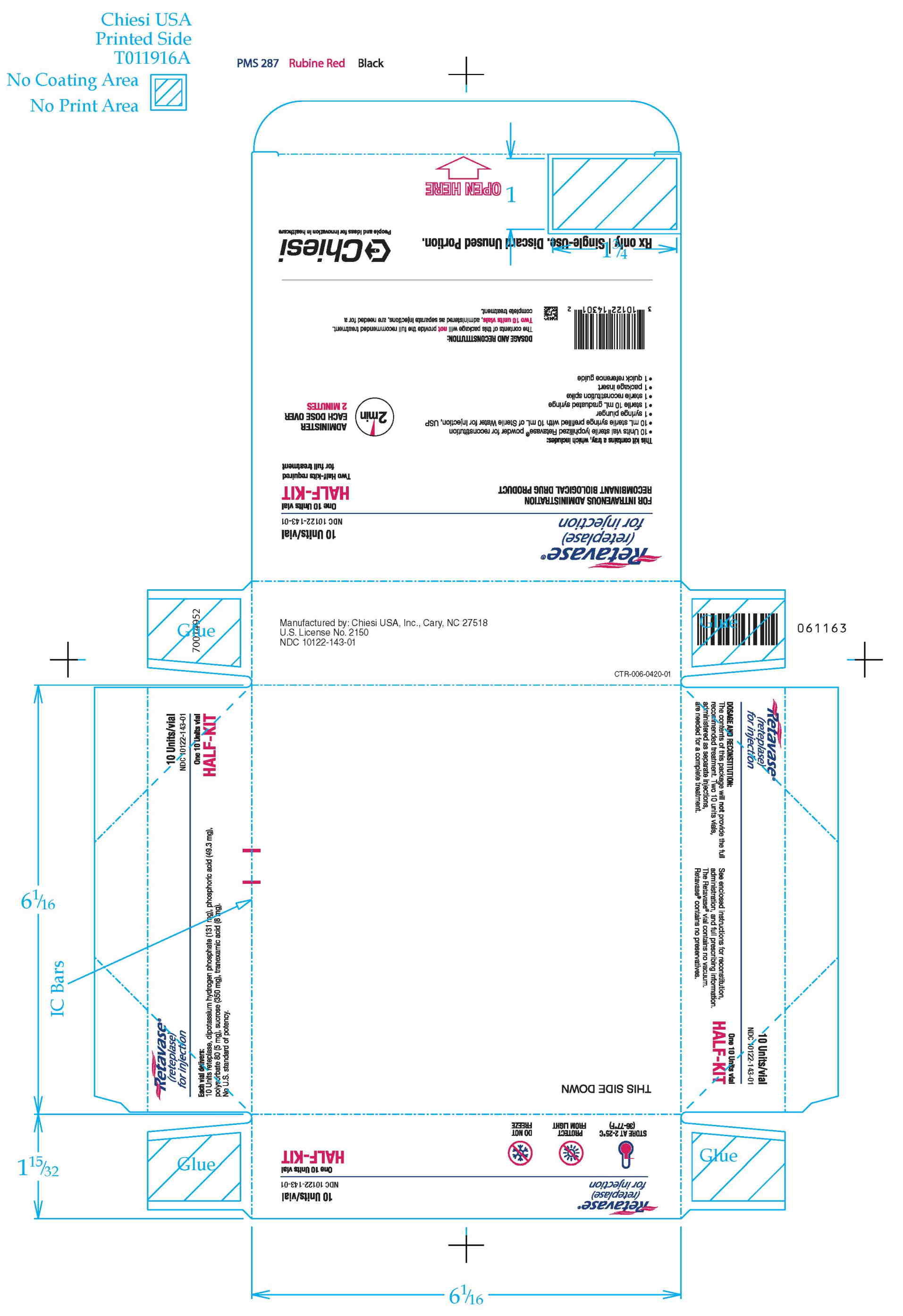
RETAVASE Half-Kit (NDC 10122-143-01): 1 single-use RETAVASE vial 10 units, 1 single-use prefilled syringe for reconstitution (10 mL Sterile Water for Injection, USP), 1 syringe plunger, 1 sterile 10 mL graduated syringe, 1 sterile reconstitution spike, 1 quick reference guide and 1 package insert.
Storage: Store RETAVASE at 2°C to 25°C (36°F to 77°F). The box should remain sealed until use to protect the lyophilisate from exposure to light.
Manufactured by:
Chiesi USA, Inc.
Cary, NC 27518
U.S. License No. 2150
Retavase® manufactured at Patheon Italia, S.p.A., Monza, Italy 20900.
To report an adverse event, record the lot number and call Medical Information at 1-888-661-9260.
RETAVASE® is a registered trademark of Chiesi USA, Inc.
The trademarks Streptase®, Activase®, and Actilyse® referenced herein are the property of their respective owners and are not affiliated with, connected to, or sponsored by Chiesi USA, Inc.
CTR-001-0422-03-SPL
Retavase Full Kit
Retavase Half Kit
| RETAVASE reteplase kit |
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
| RETAVASE reteplase kit |
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
|||||||||||||
|
Labeler — Chiesi USA, Inc.
(088084228)
Registrant — Chiesi USA, Inc. (088084228)
Frequently asked questions
- Is Activase the same as tPA?
Medical Disclaimer

Описание препарата Вазапростан® (лиофилизат для приготовления раствора для инфузий, 20 мкг) основано на официальной инструкции, утверждено компанией-производителем в 2006 году
Дата согласования: 13.07.2006
Особые отметки:
![]()
![]()
Содержание
- Действующее вещество
- ATX
- Фармакологическая группа
- Нозологическая классификация (МКБ-10)
- Состав и форма выпускa
- Фармакологическое действие
- Характеристика
- Фармакологическое действие
- Фармакокинетика
- Показания
- Противопоказания
- Применение при беременности и кормлении грудью
- Способ применения и дозы
- Побочные действия
- Взаимодействие
- Передозировка
- Меры предосторожности
- Особые указания
- Условия хранения
- Срок годности
- Аналоги (синонимы) препарата Вазапростан®
- Заказ в аптеках Москвы
Действующее вещество
ATX
Фармакологическая группа
Состав и форма выпускa
| Лиофилизат для приготовления инфузионного раствора (48,2 мг) | 1 амп. |
| алпростадил (в комплексе с альфадексом) | 20 мкг |
| вспомогательные вещества: лактоза; альфадекс |
в коробке 1 контурная ячейковая упаковка с 10 ампулами.
Фармакологическое действие
Фармакологическое действие
—
антиагрегационное, сосудорасширяющее.
Препарат ПГE1 улучшает микроциркуляцию и периферическое кровообращение, оказывает вазопротекторное действие. При системном введении вызывает расслабление гладкомышечных волокон, оказывает сосудорасширяющее действие, уменьшает ОПСС без изменения АД. При этом отмечается рефлекторное увеличение сердечного выброса и ЧСС. Способствует повышению эластичности эритроцитов, уменьшает агрегацию тромбоцитов и активность нейтрофилов, повышает фибринолитическую активность крови. Оказывает стимулирующее действие на гладкую мускулатуру кишечника, мочевого пузыря, матки; подавляет секрецию желудочного сока.
Характеристика
Лиофилизат белого цвета собран на дне ампулы, образует слой толщиной около 3 мм. После полного растворения лиофилизата в физиологическом растворе — прозрачная бесцветная жидкость.
Препарат ПГE1 улучшает микроциркуляцию и периферическое кровообращение, оказывает вазопротекторное действие. При системном введении вызывает расслабление гладкомышечных волокон, оказывает сосудорасширяющее действие, уменьшает ОПСС без изменения АД. При этом отмечается рефлекторное увеличение сердечного выброса и ЧСС. Способствует повышению эластичности эритроцитов, уменьшает агрегацию тромбоцитов и активность нейтрофилов, повышает фибринолитическую активность крови. Оказывает стимулирующее действие на гладкую мускулатуру кишечника, мочевого пузыря, матки; подавляет секрецию желудочного сока.
Фармакокинетика
ПГE1 применяется в комплексе с альфа-циклодекстрином в/в или в/а. Во время
приготовления раствора комплекс препарата распадается на составные части — ПГE1 и альфа-
циклодекстрин. При в/в введении терапевтически значимая концентрация активного вещества достигается вскоре после
начала введения препарата, а Cmax в плазме крови — в течение 2 ч от начала введения. ПГЕ
1 является эндогенным веществом с исключительно коротким T1/2 — концентрация в
плазме крови возвращается к исходному уровню за 10 с после прекращения введения. Процесс биотрансформации ПГE
1 происходит главным образом в легких, при «первом прохождении» через легкие метаболизируется 60–90%
активного вещества с образованием основных метаболитов — 15-кето — ПГE1, 15-
кето — ПГE0 и ПГE0. Основные продукты метаболизма выводятся почками
— 88% и через ЖКТ — 12% в течение 72 ч. С белками плазмы связывается 93% ПГE1.
Альфа-циклодекстрин имеет T1/2 около 7 мин, выводится почками в неизмененном виде.
Показания
Хронические облитерирующие заболевания артерий III и IV стадий (по классификации Фонтейна).
Противопоказания
Повышенная чувствительность к алпростадилу и другим компонентам препарата,
хроническая сердечная недостаточность, выраженные нарушения ритма сердца, обострение ИБС, перенесенный
инфаркт миокарда (в течение ближайших 6 мес), отек легких, инфильтративное заболевание легких,
хроническое обструктивное заболевание легких, дисфункция печени (повышенный уровень АСТ, АЛТ или ГГТ) и
заболевания печени в анамнезе, а также заболевания, сопровождающиеся повышенным риском возникновения
кровотечений и кровоизлияний (язвенная болезнь желудка или двенадцатиперстной кишки, тяжелое поражение
сосудов головного мозга, пролиферативная ретинопатия со склонностью к кровотечениям, обширная травма и
т.п.), сопутствующая терапия сосудорасширяющими и антикоагулянтными средствами, беременность, грудное
вскармливание.
Применение при беременности и кормлении грудью
Противопоказано при беременности. На время лечения следует прекратить грудное вскармливание.
Способ применения и дозы
Реклама: ООО «РЛС-Патент», ИНН 5044031277, erid=4CQwVszH9pUkpHxmQQo
В/а введение. Растворяют содержимое 1 ампулы лиофилизата Вазапростана® (соответствует 20 мкг алпростадила) в 50 мл физиологического раствора.
При отсутствии других предписаний половину содержимого ампулы Вазапростан® (соответствует 10 мкг алпростадила) вводят в/а в течение 60–120 мин при использовании устройства для инфузии. При необходимости, особенно при наличии некрозов, под строгим контролем переносимости возможно увеличение дозы до 20 мкг алпростадила (содержимое 1 ампулы). Эта дозировка обычно применяется для однократной ежедневной инфузии.
Если в/а инфузия проводится через введенный катетер, в зависимости от переносимости и тяжести заболевания, рекомендуется доза 0,1–0,6 нг/кг/мин с введением препарата в течение 12 ч при использовании устройства для инфузии (соответствует 0,25–1,5 ампулам Вазапростана).
В/в инфузия. Растворяют содержимое 2 ампул лиофилизата Вазапростана® (соответствует 40 мкг алпростадила) в 50–250 мл физиологического раствора и вводят полученный раствор в/в в течение 2 ч 2 раза в сутки или 3 ампулы (60 мкг алпростадила) в течение 3 ч 1 раз в сутки.
У пациентов с нарушенной функцией почек (почечная недостаточность при уровне креатинина более 1,5 мг/дл) в/в введение начинают с 20 мкг в течение 2 ч. При необходимости через 2–3 дня разовую дозу увеличивают до 40–60 мкг.
Для больных с почечной и сердечной недостаточностью максимальный объем вводимой жидкости — 50–100 мл/сут. Курс лечения — 4 нед.
Продолжительность лечения — в среднем 14 дней, при положительном эффекте лечение препаратом можно продолжать еще в течение 7–14 дней. При отсутствии положительного эффекта в течение 2 нед от начала лечения дальнейшее применение препарата следует прекратить.
Приготовление раствора. Готовить раствор необходимо непосредственно перед выполнением инфузии. Лиофилизат растворяется сразу после добавления физиологического раствора. В начале раствор может получиться молочно-мутным. Этот эффект создается за счет пузырьков воздуха и не имеет никакого значения. Через короткое время раствор становится прозрачным. Нельзя использовать раствор, приготовленный более 12 ч назад.
Побочные действия
Со стороны нервной системы: головная боль, судорожный синдром, головокружение, повышенная усталость, ощущение недомогания, нарушение чувствительности кожи и слизистых.
Со стороны сердечно-сосудистой системы: снижение АД, боль за грудиной, нарушения сердечного ритма, AV блокада.
Со стороны пищеварительной системы: ощущение дискомфорта в эпигастральной области, диспептические явления — диарея, тошнота, рвота.
Со стороны опорно-двигательного аппарата: гиперостоз длинных трубчатых костей (при лечении более 4 нед).
Местные реакции: боль, отек, эритема, нарушение чувствительности, флебит (проксимальнее места в/в введения).
Лабораторные показатели: лейкоцитоз, лейкопения, увеличение титра C-реактивного белка, повышение уровня трансаминаз.
Прочие: повышенное потоотделение, гипертермия, отечность конечности, в вену которой проводится инфузия.
Редко: боль в суставах, спутанность сознания, судороги центрального генеза, лихорадка, озноб, брадипноэ, артралгии, психоз, почечная недостаточность, анурия. Сообщалось о нескольких случаях развития отека легких и острой левожелудочковой недостаточности.
Крайне редко (до 1% случаев): шок, острая сердечная недостаточность, гипербилирубинемия, кровотечение, сонливость, брадипноэ, снижение функции дыхания, тахипноэ, анурия, нарушение функции почек, гипогликемия, фибрилляция желудочков сердца, AV блокада II степени, наджелудочковая аритмия, напряжение мышц шеи, повышенная раздражительность, гипотермия, гиперкапния, гиперемия кожных покровов, гематурия, перитонеальные симптомы, тахифилаксия, гиперкалиемия, тромбоцитопения, анемия.
Аллергические реакции: кожная сыпь, зуд.
Побочные эффекты, связанные с применением препарата или с самой процедурой катетеризации, исчезают после снижения дозы или прекращения инфузии.
Взаимодействие
Реклама: ООО «ВЕДАНТА», ИНН 7714886235, erid 4CQwVszH9pUkKJ7jUDd
Реклама: ООО «РЛС-Библиомед» ИНН 7714758963
Реклама: ООО «ВЕДАНТА», ИНН 7714886235, erid 4CQwVszH9pUkKJ7jUDd
Реклама: ООО «РЛС-Библиомед» ИНН 7714758963
Вазапростан® может усиливать эффект гипотензивных препаратов, вазодилататоров и антиангинальных средств. Одновременное применение Вазапростана® у пациентов, принимающих препараты, препятствующие свертыванию крови (антикоагулянты, ингибиторы агрегации тромбоцитов), может увеличить вероятность кровотечений. В комбинации с цефамандолом, цефоперазоном, цефотетаном и тромболитиками — увеличивает риск развития кровотечения. Симпатомиметики — адреналин, норадреналин, — снижают вазодилатирующий эффект.
Необходимо учитывать, что взаимодействия препаратов возможны и в том случае, если вышеперечисленные препараты применялись незадолго до того, как была начата терапия Вазапростаном®.
Передозировка
Симптомы: снижение АД, учащение ЧСС. Возможно развитие вазовагальных
реакций с бледностью кожных покровов, повышенным потоотделением, тошнотой и рвотой, что может
сопровождаться ишемией миокарда и симптомами сердечной недостаточности, а также возможны боль, отек и
покраснение ткани в месте инфузии.
Лечение: необходимо уменьшить дозу препарата или прекратить инфузию.
При выраженном снижении АД больному в положении лежа необходимо приподнять ноги. При сохранении
симптомов необходимо использовать симпатомиметики.
Меры предосторожности
С осторожностью применяют Вазапростан® при артериальной гипотензии, сердечно-сосудистой недостаточности (особое внимание следует уделять контролю нагрузки объемом раствора-носителя), у пациентов, находящихся на гемодиализе (лечение препаратом следует проводить в постдиализном периоде), у пациентов с сахарным диабетом типа 1, особенно при обширных поражениях сосудов.
Вазапростан® может влиять на способность к активному участию в уличном движении или управлению механизмами, особенно в начале лечения, при увеличении дозы и отмене препарата, а также при одновременном приеме алкоголя.
Особые указания
Алпростадил могут применять только врачи, имеющие опыт в ангиологии, знакомые с современными методами непрерывного контроля за сердечно-сосудистой системой и имеющие для этого соответствующее оборудование.
В период лечения необходим контроль АД, ЧСС, биохимических показателей крови, свертывающей системы крови (при нарушениях свертывающей системы крови или при одновременной терапии препаратами, которые влияют на свертывающую систему).
Больные ИБС, а также пациенты с периферическими отеками и почечной дисфункцией (сывороточный креатинин >1,5 мг/дл) должны находиться под наблюдением в стационаре во время лечения Вазапростаном® в течение 1 дня после прекращения его применения.
Во избежание появления симптомов гипергидратации у пациентов с почечной недостаточностью объем введенной жидкости по возможности не должен превышать 50–100 мл/сут. Обязательно динамическое наблюдение за состоянием пациента: контроль АД и ЧСС, при необходимости — контроль массы тела, баланса жидкости, измерение центрального венозного давления или проведение эхокардиографического исследования.
Флебит (проксимальнее места введения), как правило, не является причиной для прекращения терапии, признаки воспаления исчезают через несколько часов после прекращения инфузии или изменения места введения препарата, специфического лечения в подобных случаях не требуется. Катетеризация центральной вены позволяет снизить частоту проявления данного побочного действия препарата.
При повреждении ампулы лиофилизат становится влажным и клейким, и сильно уменьшается в объеме. В этом случае препарат использовать нельзя.
Условия хранения
При температуре не выше 25 °C.
Хранить в недоступном для детей месте.
Срок годности
4 года.
Не применять по истечении срока годности, указанного на упаковке.
Представленная информация о ценах на препараты не является предложением о продаже или покупке товара.
Информация предназначена исключительно для сравнения цен в стационарных аптеках, осуществляющих деятельность в
соответствии со статьей 55 Федерального закона «Об обращении лекарственных средств» от 12.04.2010 № 61-ФЗ.
Вазапростан® (Vazaprostan®) инструкция по применению
📜 Инструкция по применению Вазапростан®
💊 Состав препарата Вазапростан®
✅ Применение препарата Вазапростан®
📅 Условия хранения Вазапростан®
⏳ Срок годности Вазапростан®
Описание лекарственного препарата
Вазапростан®
(Vazaprostan®)
Основано на официальной инструкции по применению препарата, утверждено компанией-производителем
и подготовлено для электронного издания справочника Видаль 2009
года, дата обновления: 2023.06.12
Владелец регистрационного удостоверения:
Контакты для обращений:
ЮСБ ФАРМА С.А.
(Бельгия)
Код ATX:
C01EA01
(Алпростадил)
Лекарственные формы
| Вазапростан® |
Лиофилизат д/пригот. р-ра д/инф. 20 мкг: амп. 48.2 мг 10 шт. рег. №: П N013651/01 |
|
|
Лиофилизат д/пригот. р-ра д/инф. 60 мкг: амп. 10 шт. рег. №: ЛСР-001051/08 |
Форма выпуска, упаковка и состав
препарата Вазапростан®
Лиофилизат для приготовления раствора для инфузий в виде гигроскопичной лиофильной массы белого цвета.
Вспомогательные вещества: альфадекс — 646.7 мкг, лактоза безводная — 47.5 мг.
48.2 мг — ампулы стеклянные (10) — пачки картонные со специальным держателем для ампул и контролем первого вскрытия.
Лиофилизат для приготовления раствора для инфузий в виде гигроскопичной массы белого цвета.
Вспомогательные вещества: альфадекс — 1940 мкг, лактоза безводная — 47.5 мг.
49.5 мг — ампулы стеклянные (10) — пачки картонные с контролем первого вскрытия и специальным держателем для ампул.
алпростадил = ПГЕ1 (простагландин E1), МНН.
альфадекс = α-циклодекстрин (α-цд), МНН.
Фармакологическое действие
Препарат простагландина Е1 (PG E1). Улучшает микроциркуляцию и периферическое кровообращение, оказывает вазопротекторное действие.
При системном введении вызывает расслабление гладкомышечных волокон, оказывает сосудорасширяющее действие, уменьшает ОПСС без изменения АД. При этом отмечается рефлекторное увеличение сердечного выброса и ЧСС.
Способствует повышению эластичности эритроцитов, уменьшает агрегацию тромбоцитов и активность нейтрофилов, повышает фибринолитическую активность крови.
Оказывает стимулирующее действие на гладкую мускулатуру кишечника, мочевого пузыря, матки; подавляет секрецию желудочного сока.
Фармакокинетика
Применяется в комплексе с альфа-циклодекстрином в/в или в/а. Во время приготовления раствора комплекс препарата распадается на составные части — PG E1 и альфа-циклодекстрин.
Всасывание
При в/в введении терапевтически значимая концентрация активного вещества достигается вскоре после начала введения препарата, а Cmax в плазме крови достигается в течение 2 ч от начала введения.
Распределение
PG E1 связывается с белками плазмы на 93%.
Метаболизм
Процесс биотрансформации PG E1 происходит главным образом в легких, при «первом прохождении» через легкие метаболизируется 60-90% активного вещества с образованием основных метаболитов — 15-кето-PG E1, 15-кето-PG E0 и PG E0.
Выведение
PG E1 является эндогенным веществом с исключительно коротким T1/2. Концентрация в плазме крови возвращается к исходному уровню через 10 сек после прекращения введения. Выводится в виде метаболитов с мочой (88%) и через ЖКТ (12%) в течение 72 ч.
T1/2 альфа-циклодекстрина составляет около 7 мин, выводится с мочой в неизмененном виде.
Показания препарата
Вазапростан®
- хронические облитерирующие заболевания артерий III и IV стадий (по классификации Фонтейна).
Режим дозирования
Готовить раствор необходимо непосредственно перед выполнением инфузии. Лиофилизат растворяется сразу после добавления физиологического раствора. Вначале раствор может получиться молочно-мутным. Этот эффект создается за счет пузырьков воздуха и не имеет значения. Через короткое время раствор становится прозрачным.
Не допускается использование раствора, приготовленного более 12 ч назад.
Внутриартериальное введение
Лиофилизат для приготовления раствора для инфузий: 1 амп./20 мкг алпростадила
Для получения раствора для в/а введения содержимое 1 ампулы лиофилизата (соответствует 20 мкг алпростадила) следует растворить в 50 мл физиологического раствора. При отсутствии других предписаний половину ампулы Вазапростана (соответствует 10 мкг алпростадила) следует вводить в/а в течение 60-120 мин при помощи устройства для инфузии. При необходимости, особенно при наличии некрозов, под строгим контролем переносимости, дозу можно увеличить до 1 ампулы (20 мкг алпростадила). Эта доза обычно применяется для однократной ежедневной инфузии.
Если в/а введение препарата осуществляется через установленный катетер, в зависимости от переносимости и степени тяжести заболевания, рекомендуется доза 0.1-0.6 нг/кг/мин с введением препарата в течение 12 ч при использовании устройства для инфузий (соответствует 1/4-11/2 ампулам Вазапростана).
Лиофилизат для приготовления раствора для инфузий:1 амп./60 мкг алпростадила
Для получения раствора для в/а введения содержимое 1 ампулы лиофилизата (соответствует 60 мкг алпростадила) следует растворить в 50-250 мл физиологического раствора. При отсутствии других предписаний 1/6 ампулы Вазапростана (соответствует 10 мкг алпростадила) следует вводить в/а в течение 60-120 мин при помощи устройства для инфузии. При необходимости, особенно при наличии некрозов, под строгим контролем переносимости, дозу можно увеличить до 1/3 ампулы (20 мкг алпростадила). Эта доза обычно применяется для однократной ежедневной инфузии.
Если в/а введение препарата осуществляется через установленный катетер, в зависимости от переносимости и степени тяжести заболевания, рекомендуется доза 0.1-0.6 нг/кг/мин с введением препарата в течение 12 ч при использовании устройства для инфузий (соответствует 1/12—1/2 ампулам Вазапростана).
Внутривенное введение
Лиофилизат для приготовления раствора для инфузий: 1 амп./20 мкг алпростадила
Для получения раствора для в/в введения содержимое 2 ампул лиофилизата (соответствует 40 мкг алпростадила) следует растворить в 50-250 мл физиологического раствора и вводить полученный раствор в/в в течение 2 ч. Эта доза применяется 2 раза/сут.
Либо содержимое 3 ампул (соответствует 60 мкг алпростадила) растворяют в 50-250 мл физиологического раствора и вводят в/в инфузионно в течение 3 ч 1 раз/сут.
Продолжительность лечения в среднем составляет 14 дней, при положительном эффекте лечение препаратом можно продолжить еще в течение 7-14 дней. При отсутствии положительного эффекта в течение 2 недель от начала лечения дальнейшее применение препарата следует прекратить.
Лиофилизат для приготовления раствора для инфузий:1 амп./60 мкг алпростадила
Для получения раствора для в/в введения содержимое 1 ампулы лиофилизата (соответствует 60 мкг алпростадила) следует растворить в 50-250 мл физиологического раствора и вводить полученный раствор в/в в течение 3 ч 1 раз/сут.
Продолжительность лечения в среднем составляет 14 дней, при положительном эффекте лечение препаратом можно продолжить еще в течение 7-14 дней. При отсутствии положительного эффекта в течение 2 недель от начала лечения дальнейшее применение препарата следует прекратить.
У пациентов с почечной недостаточностью (сывороточный креатинин более 1.5 мг/дл) в/в введение Вазапростана необходимо начинать с 20 мкг (1 амп. 20 мкг или 1/3 амп. по 60 мкг) в течение 2 ч. При необходимости через 2-3 дня разовую дозу увеличивают до 40-60 мкг.
Для пациентов с почечной и сердечной недостаточностью максимальный объем вводимой жидкости — 50-100 мл/сут. Курс лечения — 4 недели.
Побочное действие
Со стороны ЦНС и периферической нервной системы: головная боль, головокружение, судорожный синдром, повышенная усталость, чувство недомогания, нарушение чувствительности кожи и слизистых оболочек.
Со стороны сердечно-сосудистой системы: снижение АД, боли за грудиной, нарушения сердечного ритма, AV-блокада.
Со стороны пищеварительной системы: чувство дискомфорта в эпигастрии, тошнота, рвота, диарея.
Со стороны костно-мышечной системы: гиперостоз длинных трубчатых костей (при терапии более 4 недель).
Аллергические реакции: кожная сыпь, зуд.
Местные реакции: эритема, отек, боль, нарушение чувствительности, флебит (проксимальнее места в/в введения).
Прочие: повышенное потоотделение, гипертермия, отечность конечности, в вену которой проводится инфузия.
Лабораторные показатели: лейкоцитоз, лейкопения, увеличение титра С-реактивного белка, повышение уровня трансаминаз.
Редко: артралгии, спутанность сознания, судороги центрального генеза, лихорадка, озноб, брадипноэ, психоз, почечная недостаточность, анурия. Сообщалось о нескольких случаях развития отека легких и острой левожелудочковой недостаточности.
Крайне редко (до 1% случаев): шок, острая сердечная недостаточность, гипербилирубинемия, кровотечение, сонливость, брадипноэ, снижение функции дыхания, тахипноэ, анурия, нарушение функции почек, гипогликемия, фибрилляция желудочков, AV-блокада II степени, наджелудочковая аритмия, напряжение мышц шеи, повышенная раздражительность, гипотермия, гиперкапния, гиперемия кожных покровов, гематурия, перитонеальные симптомы, тахифилаксия, гиперкалиемия, тромбоцитопения, анемия.
Побочные эффекты, связанные с применением препарата или с самой процедурой катетеризации, обратимы после снижения дозы или прекращения инфузии.
Противопоказания к применению
- хроническая сердечная недостаточность в стадии декомпенсации;
- выраженные нарушения ритма сердца;
- обострение ИБС;
- перенесенный в последние 6 мес инфаркт миокарда;
- отек легких;
- инфильтративные заболевания легких;
- хронические обструктивные заболевания легких;
- дисфункция печени (повышение уровня АСТ, АЛТ, ГГТ);
- заболевания печени в анамнезе;
- заболевания, сопровождающиеся повышенным риском возникновения кровотечений (язвенная болезнь желудка и двенадцатиперстной кишки, тяжелое поражение сосудов головного мозга, пролиферативная ретинопатия со склонностью к кровотечениям, обширная травма);
- сопутствующая терапия сосудорасширяющими и антикоагулянтными препаратами;
- беременность;
- период лактации (грудного вскармливания);
- возраст до 18 лет (эффективность и безопасность не установлены);
- повышенная чувствительность к алпростадилу и другим компонентам препарата.
С осторожностью следует назначать Вазапростан при артериальной гипотензии, сердечно-сосудистой недостаточности (обязателен контроль нагрузки объема раствора-носителя), пациентам, находящимся на гемодиализе (лечение следует проводить в постдиализном периоде), пациентам с сахарным диабетом 1 типа, особенно при обширных поражениях сосудов.
Применение при беременности и кормлении грудью
Применение Вазапростана при беременности противопоказано.
При необходимости применения препарата в период лактации следует прекратить грудное вскармливание.
Применение при нарушениях функции печени
Препарат противопоказан при нарушениях функции печени (повышение уровня АСТ, АЛТ, ГГТ), заболеваниях печени в анамнезе.
Применение при нарушениях функции почек
У пациентов с нарушениями функции почек (сывороточный креатинин более 1.5 мг/дл) в/в введение Вазапростана необходимо начинать с 20 мкг в течение 2 ч. При необходимости через 2-3 дня дозу можно увеличить до 40-60 мкг.
Для пациентов с почечной и сердечной недостаточностью максимальный объем вводимой жидкости — 50-100 мл/сут. Продолжительность курса лечения — 4 недели.
В период лечения Вазапростаном во избежание появления симптомов гипергидратации у пациентов с почечной недостаточностью объем вводимой жидкости следует ограничить до 50-100 мл/сут. Необходимо динамическое наблюдение за состоянием пациента (контроль АД и ЧСС), при необходимости – контроль массы тела, баланса жидкости, измерение центрального венозного давления или проведение эхокардиографического исследования.
Пациенты с ИБС, периферическими отеками и нарушениями функции почек (сывороточный креатинин более 1.5 мг/дл) во время лечения Вазапростаном и в течение 1 дня после прекращения применения препарата должны находиться под наблюдением в стационаре.
Применение у детей
Противопоказание: возраст до 18 лет (эффективность и безопасность не установлены).
Особые указания
Вазапростан могут применять только врачи, имеющие опыт в ангиологии, знакомые с современными методами непрерывного контроля сердечно-сосудистой системы и имеющие для этого соответствующее оборудование.
При проведении терапии Вазапростаном следует контролировать АД, ЧСС, биохимические показатели крови, показатели свертываемости крови (при нарушениях свертывающей системы крови или при одновременной терапии препаратами, которые влияют на свертывающую систему).
В период лечения Вазапростаном во избежание появления симптомов гипергидратации у пациентов с почечной недостаточностью объем вводимой жидкости следует ограничить до 50-100 мл/сут. Необходимо динамическое наблюдение за состоянием пациента (контроль АД и ЧСС), при необходимости – контроль массы тела, баланса жидкости, измерение центрального венозного давления или проведение эхокардиографического исследования.
Пациенты с ИБС, периферическими отеками и нарушениями функции почек (сывороточный креатинин более 1.5 мг/дл) во время лечения Вазапростаном и в течение 1 дня после прекращения применения препарата должны находиться под наблюдением в стационаре.
Флебит (проксимальнее места введения), как правило, не является причиной для прекращения терапии, симптомы воспаления исчезают через несколько часов после прекращения инфузии или изменения места введения препарата. Специфического лечения в подобных случаях не требуется. Катетеризация центральной вены позволяет снизить частоту проявления данного побочного действия препарата.
Влияние на способность к управлению транспортными средствами и механизмами
Вазапростан может влиять на способность к активному участию в уличном движении, к вождению автотранспорта или управлению механизмами, особенно в начале лечения, при увеличении дозы и отмене препарата, а также при одновременном приеме алкоголя.
Передозировка
Симптомы: снижение АД, увеличение ЧСС, возможно развитие вазовагальных реакций (бледность кожных покровов, повышенное потоотделение, тошнота, рвота), что может сопровождаться ишемией миокарда и симптомами сердечной недостаточности; возможны боль, отек и покраснение ткани в месте инфузии.
Лечение: необходимо уменьшить дозу препарата или прекратить инфузию. При выраженном снижении АД больному в положении лежа необходимо приподнять ноги. При сохранении симптомов необходимо использовать симпатомиметики.
Лекарственное взаимодействие
При одновременном применении Вазапростан может усиливать эффект гипотензивных средств, периферических вазодилататоров, антиангинальных препаратов.
При сочетанном применении Вазапростана с антикоагулянтами, ингибиторами агрегации тромбоцитов, цефамандолом, цефоперазоном, цефотетаном, тромболитиками повышается риск развития кровотечений.
При одновременном применении Вазапростана с эпинефрином (адреналином), норэпинефрином (норадреналином) снижается вазодилатирующий эффект.
Необходимо учитывать, что взаимодействие возможно в том случае, если вышеперечисленные препараты применялись незадолго до того, как была начата терапия Вазапростаном.
Условия хранения препарата Вазапростан®
Список Б. Препарат следует хранить в недоступном для детей месте при температуре не выше 25°С.
Срок годности препарата Вазапростан®
Срок годности — 4 года.
При повреждении ампулы лиофилизат становится влажным и клейким, сильно уменьшается в объеме. В этом случае использовать препарат нельзя.
Условия реализации
Препарат отпускается по рецепту.
Контакты для обращений
ЮСБ ФАРМА С.А.
(Бельгия)

|
|
ЮСБ Фарма ООО 105082 Москва, Переведеновский пер. 13, стр. 21 |
Если вы хотите разместить ссылку на описание этого препарата — используйте данный код
Вазапростан — инструкция по применению
Синонимы, аналоги
Статьи
Регистрационный номер
ЛСР-001051/08
Торговое наименование препарата
Вазапростан®
Международное непатентованное наименование
Алпростадил
Лекарственная форма
лиофилизат для приготовления раствора для инфузий
Состав
1 ампула содержит:
Действующее вещество: алпростадил (клатратный комплекс с альфадексом 1:1)60 мкг;
Вспомогательные вещества: альфадекс (α-циклодекстрин) 1940,0 мкг, лактозы безводной 47,5 мг.
Алпростадил = ПГЕ1 (простагландин Е1), МНН
Альфадекс = α-циклодекстрин (α-цд), МНН
Описание
Гигроскопичная лиофильная масса белого цвета.
Фармакотерапевтическая группа
Вазодилатирующее средство — простагландина Е1, аналог синтетический
Код АТХ
G04BE01
Фармакодинамика:
Препарат простагландина Е1 (PgE1) улучшает микроциркуляцию и периферическое кровообращение, оказывает вазопротекторное действие. При системном введении вызывает расслабление гладкомышечных волокон, оказывает сосудорасширяющее действие, уменьшает общее периферическое сосудистое сопротивление без изменения артериального давления. При этом отмечается рефлекторное увеличение сердечного выброса и частоты сердечных сокращений. Способствует повышению эластичности эритроцитов, уменьшает агрегацию тромбоцитов и активность нейтрофилов, повышает фибринолитическую активность крови. Оказывает стимулирующее действие на гладкую мускулатуру кишечника, мочевого пузыря, матки; подавляет секрецию желудочного сока.
Фармакокинетика:
Применяется PgE1 в комплексе с альфациклодекстрином внутривенно или внутриартериально. Во время приготовления раствора комплекс препарата распадается на составные части — PgE1 и альфациклодекстрин. При внутривенном введении терапевтически значимая концентрация активного вещества достигается вскоре после начала введения препарата, а максимальная концентрация в плазме крови достигается в течение 2 часов от начала введения. PgЕ1 является эндогенным веществом с исключительно коротким периодом полувыведения — концентрация в плазме крови возвращается к исходному уровню через 10 секунд после прекращения введения препарата. Процесс биотрансформации PgE1 происходит главным образом в легких, при «первом прохождении» через легкие метаболизируется 60-90% активного вещества с образованием основных метаболитов — 15-KeTO-PgE1 15-KeTO-PgE0 и PgE0. Выводятся основные продукты метаболизма почками — 88% и через желудочно-кишечный тракт — 12% в течение 72 часов. С белками плазмы крови связывается 93% PgE1. Альфациклодекстрин имеет период полувыведения около 7 минут, выводится почками в неизмененном виде.
Показания:
Хронические облитерирующие заболевания артерий III и IV стадий (по классификации Фонтейна) у пациентов, которым невозможно провести реваскуляризацию или после неудачной реваскуляризации.
Внутривенное применение при лечении хронических облитерирующих заболеваний артерий IV стадии не рекомендуется.
Противопоказания:
- Повышенная чувствительность к алпростадилу или другим компонентам препарата;
- Хроническая сердечная недостаточность III-IV функционального класса по NYHA;
- Гемодинамически значимые нарушения ритма сердца;
- Обострение течения ишемической болезни сердца, перенесенный в последние шесть месяцев инфаркт миокарда;
- Митральный и/или аортальный стеноз и/или недостаточность;
- Острый отек легких или наличие отека легких в анамнезе у пациентов с сердечной недостаточностью;
- Тяжелая степень хронической обструктивной болезни легких или легочное веноокклюзионное заболевание;
- Инфильтративное заболевание легких;
- Заболевания, сопровождающиеся повышенным риском возникновения кровотечений (язвенная болезнь желудка и/или двенадцатиперстной кишки;
- Сопутствующая терапия сосудорасширяющими или антикоагулянтными лекарственными средствами;
- Нарушение мозгового кровообращения в течение последних 6 месяцев;
- Тяжелая артериальная гипотензия;
- Нарушения функции почек (олигурия);
- Острая печеночная недостаточность (повышение активности аспартатаминотрансферазы, аланинаминотрансферазы или гамма-глутамилтрансферазы) или тяжелая печеночная недостаточность, в том числе в анамнезе;
- Общие противопоказания для инфузионной терапии (например, хроническая сердечная недостаточность, отек и гипергидратация легких или мозга);
- Беременность и период грудного вскармливания.
- Возраст до 18 лет (эффективность и безопасность не установлены).
С осторожностью:
Артериальная гипотензия.
Хроническая сердечная недостаточность I-II функционального класса по NYHA (особое внимание следует уделять контролю нагрузки объема раствора-носителя). Пациенты находящиеся на гемодиализе (лечение препаратом следует проводить в постдиализном периоде).
Пациенты с сахарным диабетом типа 1, особенно при обширных поражениях сосудов (у пациентов пожилого возраста).
Беременность и лактация:
Женщины репродуктивного возраста
Женщинам репродуктивного возраста следует использовать надежные методы контрацепции во время лечения.
Беременность и период лактации
Применение препарата Вазапростан® при беременности противопоказано. При необходимости назначения в период лактации, грудное вскармливание следует прекратить.
Фертильность
По данным доклинических исследований алпростадил не оказывает никаких нежелательных реакций со стороны фертильность.
Способ применения и дозы:
Вводить только внутривенно или внутриартериально. Внутривенное применение при лечении хронических облитерирующих заболеваний артерий IV стадии не рекомендуется.
Готовить раствор необходимо непосредственно перед проведением инфузии. Лиофилизат растворяется сразу после добавления изотонического 0,9 % раствора для инъекций натрия хлорида. В начале раствор может получиться молочномутным. Этот эффект создается за счет пузырьков воздуха. Через короткое время раствор становится прозрачным. Нельзя использовать раствор по истечении 12 часов после приготовления.
Применение в педиатрии:
Вазапростан® не рекомендуется назначать детям и подросткам в возрасте до 18 лет, так как безопасность и эффективность препарата в этих возрастных группах не изучались.
Внутриартериальное введение III и IV стадия
Растворить содержимое одной ампулы препарата Вазапростан® (соответствует 60 мкг алпростадила) в 50-250 мл изотонического 0,9 % раствора для инъекций натрия хлорида.
При отсутствии других предписаний содержимое 1/6 ампулы препарата Вазапростан® (соответствует 10 мкг алпростадила) вводить внутриартериально в течение 60-120 мин. при использовании устройства для инфузии. При необходимости, особенно при наличии некроза, под строгим контролем переносимости препарата, дозу можно увеличить до 1/3 ампулы (соответствует 20 мкг алпростадила). Эта дозировка обычно применяется для однократной ежедневной инфузии.
Внутривенная инфузия III стадия
Растворить содержимое одной ампулы препарата Вазапростан® (соответствует 60 мкг алпростадила) в 50-250 мл изотонического 0,9 % раствора для инъекций натрия хлорида и вводить приготовленный раствор инфузионно внутривенно в течение 2 часов 1 раз в день.
У пациентов с нарушенной функцией почек (концентрация креатинина в сыворотке крови более 1,5 мг/дл) внутривенное введение препарата Вазапростан® начинают с 20 мкг (1/3 ампулы), вводя его в течение двух часов. При необходимости через 2-3 дня разовую дозу 20 мкг увеличивают до 40-60 мкг.
Пациентам с почечной недостаточностью или пациентам, которых можно отнести к группе риска в связи с нарушением функции сердца, объем вводимой при инфузии жидкости следует ограничить до 50-100 мл в сутки во избежание появления симптомов гипергидратации. Инфузию следует проводить с помощью инфузионных насосов.
Продолжительность терапии составляет в среднем — 14 дней, при положительном терапевтическом эффекте лечение препаратом можно продолжить еще в течение 7-14 дней. Курс лечения не должен превышать 3-4 недели. При отсутствии положительного эффекта в течение 2 недель от начала лечения дальнейшее применение препарата следует прекратить.
Побочные эффекты:
Возможные побочные эффекты приведены ниже по системам организма и частоте возникновения: очень часто (>1/10), часто (>1/100, <1/10), нечасто (>1/1000, <1/100), редко (>1/10000, <1/1000), очень редко (<1/10000), неизвестно (невозможно оценить по доступным данным).
При использовании препарата Вазапростан® наблюдаются следующие нежелательные эффекты:
Нарушение со стороны кроветворной и лимфатической системы:
Редко: тромбоцитопения, лейкопения, лейкоцитоз;
Очень редко: кровотечение, анемия.
Нарушения со стороны нервной системы:
Часто: головная боль;
Редко: спутанность сознания, судороги центрального генеза;
Очень редко: сонливость, головокружение;
Неизвестно: нарушение мозгового кровообращения.
Нарушения со стороны сердца:
Нечасто: снижение систолического артериального давления, тахикардия, стенокардия;
Редко: аритмия, бивентрикулярная сердечная недостаточность, отёк лёгких;
Очень редко: острая сердечная недостаточность, фибрилляция желудочков сердца, атриовентрикулярная блокада II степени, наджелудочковая аритмия;
Неизвестно: инфаркт миокарда.
Нарушения со стороны сосудов:
Очень редко: шок.
Нарушения со стороны дыхательной системы, органов грудной клетки и средостения:
Редко: отек легких;
Очень редко: брадипноэ, снижение функции дыхания, тахипноэ, гиперкапния;
Неизвестно: диспноэ.
Нарушения со стороны желудочно-кишечного тракта:
Нечасто: диарея, тошнота, рвота;
Очень редко: перитонеальные симптомы.
Нарушения со стороны обмена веществ и питания:
Очень редко: гипогликемия, гиперкалиемия.
Нарушения со стороны печени и желчевыводящих путей:
Редко: повышение активности «печеночных» трансаминаз, повышение активности гамма-глутамилтрансферазы;
Очень редко: гипербилирубинемия.
Нарушения со стороны почек и мочевыводящих путей:
Очень редко: нарушение функции почек, гематурия.
Нарушения со стороны кожи и подкожных тканей:
Часто: покраснение, отёк, «прилив» крови.
Аллергические реакции:
Нечасто: кожная гиперчувствительность, например, сыпь.
Нарушения со стороны скелетно-мышечной и соединительной ткани:
Очень редко: напряжение мышц шеи, при длительном применении (4 недели и более) может возникать обратимый гиперостоз трубчатых костей, также возможно увеличение титра С-реактивного белка;
Нечасто: боль в суставах.
Общие расстройства и нарушения в месте введения:
Часто: боль, головная боль, после внутриартериального введения: ощущение жара, ощущение распирания, локальный отёк, парестезия;
Нечасто: после внутривенного введения: ощущение жара, ощущение распирания, локальный отёк, парестезия, лихорадка, повышенное потоотделение, озноб;
Очень редко: анафилаксия/ анафилактоидная реакция, утомляемость, общее недомогание, повышенная раздражительность, гипотермия, тахифилаксия;
Неизвестно: флебит на участке инъекции, тромбоз, кровотечение в месте введения катетера.
Передозировка:
Передозировка препаратом Вазапростан® может проявляться выраженным снижением артериального давления и рефлекторной тахикардией. Возможно развитие вазовагальных реакций с бледностью кожных покровов, повышенным потоотделением, тошнотой и рвотой. Возможны боль, отек и покраснение кожи в месте инфузии.
При симптомах передозировки необходимо уменьшить дозу препарата Вазапростан® или прекратить инфузию. При значительном снижении артериального давления пациенту в положении «лежа» необходимо приподнять ноги. При сохранении симптомов необходимо использовать симпатомиметики.
Взаимодействие:
Алпростадил может усиливать эффект гипотензивных средств, вазодилататоров. Одновременное применение алпростадила у пациентов, принимающих антикоагулянты и/или антиагреганты может увеличить вероятность кровотечения.
Симпатомиметики эпинефрин (адреналин), норэпинефрин (норадреналин) снижают вазодилатирующий эффект. Необходимо учитывать, что взаимодействия препаратов возможны и в том случае, если вышеперечисленные средства применялись незадолго до того, как была начата терапия препаратом Вазапростан®.
Особые указания:
Вазапростан® нельзя вводить болюсно!
Вазапростан® могут применять только врачи, имеющие опыт работы в ангиологии, знакомые с современными методами непрерывного контроля за показателями сердечно-сосудистой системы.
Пациенты с ишемической болезнью сердца, а также пациенты с периферическими отеками и нарушением функции почек (сывороточный креатинин более 1,5 мг/дл) должны находиться под наблюдением в стационаре в течение одного дня после прекращения применения препарата.
Пациентам с почечной недостаточностью или пациентам, которых можно отнести к группе риска в связи с нарушением функции сердца, объем вводимой при инфузии жидкости следует ограничить до 50-100 мл в сутки во избежание появления симптомов гипергидратации.
В период лечения необходим регулярный контроль артериального давления, частоты сердечных сокращений, биохимических показателей крови, свертывающей системы крови (при нарушениях свертывающей системы крови или при одновременной терапии средствами, которые влияют на показатели свертывающей системы), при необходимости — контроль массы тела, баланса жидкости, измерение центрального венозного давления, проведение эхокардиографического исследования.
Флебит (проксимальнее места введения), как правило, не является причиной для прекращения терапии, признаки воспаления исчезают через несколько часов после прекращения инфузии или изменения места введения препарата, специфического лечения в подобных случаях не требуется. Катетеризация центральной вены позволяет снизить частоту проявления данного побочного действия препарата.
При повреждении ампулы лиофилизат становится влажным и клейким, и сильно уменьшается в объеме. В этом случае препарат использовать нельзя.
Влияние на способность управлять транспортными средствами и механизмами:
Алпростадил может вызывать снижение систолического артериального давления и, тем самым, снижает способность к управлению транспортными средствами и занятию другими потенциально опасными видами деятельности, требующими повышенной концентрации внимания и быстроты психомоторных реакций.
Форма выпуска/дозировка:
Лиофилизат для приготовления раствора для инфузий 60 мкг.
Упаковка:
По 49,5 мг (соответствует 60 мкг алпростадила) в ампулы нейтрального стекла с синей точкой над местом надпила и маркировочным кольцом синего цвета.
По 10 ампул вместе с инструкцией по применению в пачку картонную со специальным держателем для ампул.
В случае упаковки препарата па ЗАО «ФирмФирма «Сотекс»
По 5 ампул помещают в контурную ячейковую упаковку из пленки поливинилхлоридной и пленки полимерной или без пленки полимерной.
По 2 контурные ячейковые упаковки по 5 ампул вместе с инструкцией по применению помещают в пачку из картона.
Условия хранения:
При температуре не выше 25 °С, в недоступном для детей месте.
Срок годности:
4 года.
Не применять по истечении срока годности!
Условия отпуска
По рецепту
Производитель
ЮСБ Фарма ГмбХ, Alfred-Nobel-Strasse 10, 40789 Monheim, Germany, Германия
Владелец регистрационного удостоверения/организация, принимающая претензии потребителей:
Шварц Фарма АГ
Комментарии
(видны только специалистам, верифицированным редакцией МЕДИ РУ)

Внешний вид упаковки может отличаться от фотографии
Оплата и способы получения
в Москве
Оплата и способы получения
в Москве
Самовывоз
Завтра или позже бесплатно из 3519 аптек
Оплата картой или наличными в аптеке
Доставка
Конкретный срок и стоимость доставки зависят от вашего адреса
Информация о товаре
Форма выпуска:
раствор для подкожного введения
Количество в упаковке:
2 шт.
Производитель:
Sanofi-Winthrop
Страна:
Франция
Страна производства может отличаться, проверяйте при получении заказа
Инструкция на Кевзара 175 мг/мл, раствор для подкожного введения, 1,14 мл, 2 шт.
Состав
| Ингредиент | Количество (дозировка 150 мг) |
Количество (дозировка 200 мг) |
||
| в 1 мл | в шприце/шприц-ручке | в 1 мл | в шприце/шприц-ручке | |
| Действующее вещество | ||||
| Сарилумаб | 131,6 мг | 150 мг | 175 мг | 200 мг |
| Вспомогательные вещества | ||||
| L-гистидин L-гистидина гидрохлорида моногидрат |
3,26 мг1 | 3,71 мг1 | 3,26 мг1 | 3,71 мг1 |
| L-аргинина гидрохлорид |
7,84 мг2 | 8,94 мг2 | 7,84 мг2 | 8,94 мг2 |
| Сахароза | 50 мг | 57 мг | 50 мг | 57 мг |
| Полисорбат-20 | 2 мг | 2,28 мг | 2 мг | 2,28 мг |
| Вода для инъекций | до 1,00 мл | до 1,14 мл | до 1,00 мл | до 1,14 мл |
1) Содержание L-гистидина и L-гистидина гидрохлорида моногидрата, приведено в пересчете на L-гистидин.
2) Содержание L-аргинина гидрохлорида приведено в пересчете на L-аргинин.
Описание
Прозрачная или опалесцирующая, бесцветная или коричневато-желтого цвета жидкость.
Фармакотерапевтическая группа
Иммунодепрессанты, ингибиторы интерлейкина.
Фармакодинамика
Механизм действия
Сарилумаб – человеческое моноклональное антитело (подтип IgGl) к рецептору интерлейкина-6 (ИЛ-6). Сарилумаб специфически связывается как с растворимыми, так и с мембранными рецепторами ИЛ-6 (IL-6Rα), и подавляет ИЛ-6-опосредованную передачу сигнала с вовлечением убиквитарного сигнального белка гликопротеина 130 (gp130) и STAT-3 белков (трансдукторы сигналов и активаторы транскрипции-3).
В функциональных исследованиях на человеческих клетках было показано, что сарилумаб способен блокировать сигнальный путь ИЛ-6, измеряемый по степени подавления STAT-3 белков, только в присутствии ИЛ-6.
ИЛ-6 – это плейотропный цитокин, который стимулирует различные клеточные ответы, такие как пролиферация, дифференцировка, выживаемость и апоптоз клеток; активирует высвобождение белков острой фазы воспаления в гепатоцитах, включая С-реактивный белок (СРБ) и сывороточный амилоид А. Повышенный уровень ИЛ-6, выявляемый в синовиальной жидкости у пациентов с ревматоидным артритом, играет важную роль как в развитии патологического воспаления, так и в развитии деструкции суставов, которые являются отличительными признаками ревматоидного артрита. ИЛ-6 участвует в различных физиологических процессах, таких как миграция и активация Т- и В-лимфоцитов, моноцитов и остеокластов, приводя к развитию системного воспаления, воспаления синовиальной оболочки суставов и развитию костных эрозий у пациентов с ревматоидным артритом. Действие сарилумаба приводит к уменьшению воспаления и сопровождается изменениями лабораторных показателей, такими как снижение абсолютного числа нейтрофилов (АЧН) и повышение концентрации липидов.
Фармакодинамические эффекты
После подкожного введения сарилумаба в разовых дозах 150 мг и 200 мг у пациентов с ревматоидным артритом наблюдалось быстрое снижение уровня СРБ. Уровень СРБ снижался до нормальных значений уже через 4 дня после начала лечения. У пациентов с ревматоидным артритом после введения разовой дозы сарилумаба АЧН снижалось до минимального значения в течение 3-4 дней, а затем восстанавливалось до исходного уровня. Лечение сарилумабом приводило к снижению уровня фибриногена и сывороточного амилоида А, а также к повышению уровней гемоглобина и альбумина сыворотки крови.
Клиническая эффективность и безопасность
Эффективность и безопасность препарата Кевзара® были оценены в 3-х рандомизированных двойных слепых контролируемых многоцентровых исследованиях.
Плацебо-контролируемые исследования
В исследовании MOBILITY принимали участие 1197 пациентов с ревматоидным артритом с недостаточным клиническим ответом на терапию метотрексатом. Пациенты получали препарат Кевзара® в дозах 200 мг, 150 мг или плацебо каждые 2 недели одновременно с метотрексатом. В исследовании TARGET принимали участие 546 пациентов с ревматоидным артритом с недостаточным клиническим ответом на терапию одним или несколькими антагонистами ФНО-α или в случае их непереносимости. Пациенты получали препарат Кевзара® в дозах 200 мг или 150 мг или плацебо в сочетании с традиционными болезнь-модифицирующими антиревматическими препаратами [тБМАРП] каждые 2 недели.
• Клинический ответ
На 24-й неделе терапии в обоих исследованиях у пациентов, получавших препарат Кевзара® в дозе 200 мг или 150 мг в сочетании с тБМАРП 1 раз каждые 2 недели частота ответа ACR20, ACR50 и ACR70 была выше, чем у пациентов, получавших плацебо. В открытой продленной фазе исследования эти результаты сохранялись в течение 3-х лет терапии.
В исследовании MOBILITY к 52-й неделе большая часть пациентов, получавших препарат Кевзара в дозе 200 мг или 150 мг 1 раз каждые 2 недели в сочетании с метотрексатом, достигла ремиссии, определяемой по DAS28-CPБ<2,6 (Индексу активности болезни по 28 суставам – С-реактивный белок), по сравнению с группой пациентов, получавших плацебо в сочетании с метотрексатом.
В исследованиях MOBILITY и TARGET в группе активного лечения более высокая частота ответа по критериям ACR20 по сравнению с группой плацебо наблюдалась уже через 2 недели; результаты сохранялись на протяжении всего исследования.
Результаты исследования MOBILITY через 52 недели терапии были сходны с результатами исследования TARGET через 24 недели.
• Рентгенологический ответ
В исследовании MOBILITY эффективность обеих доз препарата Кевзара® в сочетании с метотрексатом превосходила эффективность комбинации плацебо и метотрексата в отношении структурных повреждений суставов, оцениваемых по изменению модифицированного счёта Шарпа/ван дер Хейде по сравнению с исходным уровнем через 24 и через 52 недели.
Через 52 недели терапии препаратом Кевзара® в дозе 200 мг и в дозе 150 мг в сочетании с метотрексатом было отмечено уменьшение прогрессирования структурных повреждений на 91% и 68%, соответственно, по сравнению с комбинацией плацебо и метотрексата.
• Изменения функционального статуса
В исследованиях MOBILITY и TARGET к 16-й и 12-й неделе неделе терапии соответственно было продемонстрировано более выраженное улучшение функционального статуса по HAQ-DI в группах препарата Кевзара® по сравнению с плацебо, которое сохранялось до 52 недели в исследовании MOBILITY.
Исследование с использованием активного препарата в качестве контроля
Исследование MONARCH – 24-недельное рандомизированное двойное слепое, двойное маскированное исследование, в котором сравнивали монотерапию препаратом Кевзара® в дозе 200 мг с монотерапией адалимумабом в дозе 40 мг.
Препарат Кевзара® в дозе 200 мг превосходил адалимумаб в дозе 40 мг в отношении снижения активности заболевания и улучшения функционального статуса.
Фармакокинетика
Фармакокинетика сарилумаба исследовалась у 2186 пациентов с ревматоидным артритом, из которых 751 пациент получал препарат Кевзара® в дозе 150 мг и 891 пациент – в дозе 200 мг подкожно 1 раз каждые 2 недели в течение до 52-х недель. Медиана максимальной концентрации достигалась через 2-4 дня после введения препарата.
Всасывание
В равновесном состоянии концентрация сарилумаба в интервалах между введениями, которая измерялась с помощью площади под кривой зависимости концентрации от времени (AUC), увеличивалась в 2 раза при повышении дозы со 150 мг до 200 мг при введении 1 раз каждые 2 недели. Равновесное состояние достигалось через 12-16 недель с 2-3-кратным накоплением по сравнению с концентрацией после введения разовой дозы.
При введении дозы 150 мг 1 раз каждые 2 недели расчетные средние значения (±стандартное отклонение) в равновесном состоянии AUC, минимальная концентрация (Cmin) и максимальная концентрация (Сmax) сарилумаба составили 210±115 мг·сут/л, 6,95±7,6 мг/л и 20,4±8,27 мг/л, соответственно.
При введении дозы 200 мг 1 раз каждые 2 недели расчетные средние значения (±стандартное отклонение) в равновесном состоянии AUC, Cmin и Сmax сарилумаба составили 396±194 мг·сут/л, 16,7±13,5 мг/л и 35,4±13,9 мг/л, соответственно.
Распределение
У пациентов с ревматоидным артритом кажущийся объем распределения в равновесном состоянии составил 8,3 л.
Метаболизм
Метаболизм сарилумаба не изучен. Предполагается, что сарилумаб, как и другие моноклональные антитела, распадается на небольшие пептиды и аминокислоты через катаболизм таким же образом, как и эндогенный иммуноглобулин (IgG).
Выведение
Выведение сарилумаба происходит одновременно двумя путями: линейным и нелинейным. При высоких концентрациях выведение осуществляется преимущественно посредством линейного ненасыщаемого протеолитического пути, в то время как при более низких концентрациях преобладает нелинейное, насыщаемое, опосредованное мишенями, выведение. Эти параллельные пути определяют начальный период полувыведения от 8 до 10 дней и терминальный период полувыведения, зависящий от концентрации, от 2 до 4 дней.
После достижения равновесного состояния при введении последней дозы сарилумаба 150 мг и 200 мг медиана времени до неопределяемых концентраций, составляет 30 и 49 дней, соответственно. Моноклональные антитела не выводятся почками и печенью.
Линейность/нелинейность
У пациентов с ревматоидным артритом наблюдалась более чем дозозависимое увеличение фармакокинетической экспозиции. В равновесном состоянии концентрация в перерывах между введениями препарата измерялась AUC, которая увеличивалась примерно в 2 раза с повышением дозы в 1,33 раза от 150 до 200 мг при введении препарата 1 раз каждые 2 недели.
Взаимодействие с субстратами цитохрома Р450
Симвастатин является субстратом изофермента CYP3A4 и транспортного белка ОАТР1В1. У 17 пациентов с ревматоидным артритом через неделю после разового подкожного введения сарилумаба в дозе 200 мг, экспозиция симвастатина и симвастатиновой кислоты уменьшилась на 45% и 36% соответственно.
Особые группы пациентов
Возраст, пол, этническая принадлежность и масса тела
Популяционный анализ фармакокинетики у взрослых пациентов с ревматоидным артритом (возраст от 18 до 88 лет; 14% пациентов в возрасте старше 65 лет), показал, что возраст, пол и этническая принадлежность не оказывают значимого влияния на фармакокинетику сарилумаба. У пациентов с массой тела более 100 кг применение сарилумаба в обеих дозах 150 мг и 200 мг продемонстрировало эффективность; однако пациенты с массой тела более 100 кг получили большую терапевтическую пользу при применении дозы 200 мг.
Нарушения функции почек
Каких-либо официальных исследований влияния почечной недостаточности на фармакокинетику сарилумаба не проводилось. Почечная недостаточность легкой и средней степени тяжести не влияет на фармакокинетику сарилумаба. Пациентам с почечной недостаточностью легкой и средней степени тяжести коррекция дозы не требуется. Применение сарилумаба у пациентов с почечной недостаточностью тяжелой степени не изучалось.
Нарушения функции печени
Каких-либо официальных исследований влияния печеночной недостаточности на фармакокинетику сарилумаба не проводилось.
Кевзара: Показания
Препарат Кевзара® в комбинации с метотрексатом показан для лечения ревматоидного артрита умеренной или высокой степени активности у взрослых пациентов при недостаточном ответе на терапию одним или несколькими болезнь-модифицирующими антиревматическими препаратами (БМАРП) или при их непереносимости.
Препарат Кевзара® может назначаться в монотерапии при непереносимости метотрексата или при нецелесообразности терапии метотрексатом.
Способ применения и дозы
Лечение препаратом Кевзара® должно назначаться и проводиться под контролем специалистов, имеющих опыт диагностики и лечения ревматоидного артрита.
Рекомендуемая доза препарата Кевзара® составляет 200 мг 1 раз каждые 2 недели. Препарат вводят подкожно.
При развитии нейтропении, тромбоцитопении, повышении активности «печеночных» ферментов рекомендуется уменьшить дозу с 200 мг 1 раз каждые 2 недели до 150 мг 1 раз каждые 2 недели.
Пропуск дозы
Если введение препарата Кевзара® пропущено, и с момента пропуска введения препарата прошло 3 дня или менее, то следующую дозу необходимо ввести как можно скорее. Следующую очередную дозу вводят в обычное запланированное время.
Если с момента пропуска введения препарата прошло 4 дня или более, то следующую дозу вводят в следующее обычное запланированное время. При этом дозу нельзя удваивать.
Особые группы пациентов
Нарушения функции пачек
У пациентов с почечной недостаточностью легкой и средней степени тяжести коррекции дозы препарата не требуется. Применение препарата Кевзара® у пациентов с почечной недостаточностью тяжелой степени не изучалось.
Нарушения функции печени
Безопасность и эффективность препарата Кевзара® не изучались у пациентов с нарушениями функции печени, включая пациентов с положительными результатами серологических исследований на вирус гепатита В (HBV) или вирус гепатита С (HCV).
Пожилые пациенты
У пациентов в возрасте старше 65 лет коррекции дозы не требуется.
Дети
Не следует применять препарат Кевзара® у детей в возрасте до 18 лет (безопасность и эффективность препарата при ревматоидном артрите не установлены).
Препарат Кевзара® вводят подкожно.
Все содержимое (1,14 мл) предварительно заполненного шприца/предварительно заполненной шприц-ручки следует вводить подкожно. Места инъекций (область живота, наружная поверхность бедра, наружная поверхность плеча) следует чередовать при каждой инъекции. Не следует вводить препарат в болезненную и поврежденную кожу, в места с кровоподтеками и рубцами.
Пациент может самостоятельно выполнять подкожную инъекцию препарата Кевзара®, или ее может выполнять лицо, осуществляющее уход за пациентом. Пациент или лицо, осуществляющее уход за пациентом, до начала применения препарата Кевзара® должны быть обучены подготовке и введению препарата.
Применение при беременности и кормлении грудью
Женщины детородного возраста
Женщины детородного возраста должны использовать эффективные методы контрацепции во время терапии препаратом Кевзара® и в течение 3-х месяцев после ее окончания.
Беременность
Данные по применению сарилумаба у беременных женщин ограничены или отсутствуют. Известно, что моноклональные антитела проникают через плаценту, при этом большее количество антител проникает через плаценту в III триместре.
Исследования на животных не дают прямых или косвенных указаний на негативное влияние сарилумаба с точки зрения репродуктивной токсичности. Препарат Кевзара® не следует применять во время беременности, за исключением тех случаев, когда потенциальная польза применения для матери превышает потенциальный риск для плода.
Период грудного вскармливания
Неизвестно, экскретируется ли сарилумаб в грудное молоко или подвергается ли системной абсорбции у новорожденного после грудного вскармливания. Информация относительно влияния сарилумаба на ребенка, находящегося на грудном вскармливании, или продукцию грудного молока отсутствует. Поскольку IgGl в небольших количествах могут экскретироваться в грудное молоко, следует с учетом пользы грудного вскармливания для ребенка и пользы терапии для женщины принять решение либо о прекращении грудного вскармливания, либо об отмене сарилумаба.
Фертильность
Данные о воздействии сарилумаба на фертильность у людей отсутствуют. Исследования на животных показали отсутствие негативного влияния на фертильность самок и самцов.
Кевзара: Противопоказания
- Повышенная чувствительность к активному веществу или любому вспомогательному компоненту препарата.
- Активные серьезные инфекционные заболевания (см. раздел «Особые указания»).
- Дети в возрасте до 18 лет в связи с неустановленными эффективностью и безопасностью у детей с ревматоидным артритом.
С осторожностью
- У пациентов с хронической или рецидивирующей инфекцией; серьезными или оппортунистическими инфекциями в анамнезе; с сопутствующими заболеваниями, предрасполагающими к развитию инфекций; после контакта с больными туберкулезом; проживавших или посещавших регионы, эндемичные по туберкулезу или микозам (необходимо оценить соотношение пользы и риска перед началом применения, см. раздел «Особые указания»).
- У пациентов с ВИЧ-инфекцией;
- У пациентов с повышенным риском развития перфорации желудочно-кишечного тракта (см. раздел «Особые указания»).
- У пациентов пожилого возраста (в связи с более высокой частотой развития инфекций у данной категории пациентов, см. раздел «Особые указания»).
- Ограничения по применению препарата в зависимости от возраста пациента приведены в разделе «Способ применения и дозы».
Кевзара: Побочные действия
Наиболее частыми нежелательными реакциями, которые наблюдались в клинических исследованиях, были нейтропения, повышение активности АЛТ, эритема в месте инъекции, инфекции верхних отделов дыхательных путей, инфекции мочевыводящих путей.
Наиболее частыми серьезными нежелательными реакциями были инфекции.
Безопасность препарата Кевзара® в сочетании с БМАРП оценивалась на основании данных 7 клинических исследований, 2 из которых были плацебо-контролируемыми, с включением 2887 пациентов (выборка для оценки долгосрочной безопасности). Из них 2170 пациентов получали препарат Кевзара® не менее 24 недель, 1546 пациентов – не менее 48 недель, 1020 пациентов – не менее 96 недель и 624 пациента – не менее 144 недель.
Частота нежелательных реакций, перечисленных ниже, определялась следующим образом: очень часто (≥1/10); часто (от ≥1/100 до <1/10); нечасто (от ≥1/1000 до <1/100); редко (от ≥1/10000 до <1/1000); очень редко (<1/10000). В пределах каждой частотной группы нежелательные реакции представлены в порядке уменьшения серьезности.
Инфекционные и паразитарные заболевания: часто – инфекции верхних отделов дыхательных путей, инфекции мочевыводящих путей, назофарингит, герпес ротовой полости.
Нарушения со стороны крови и лимфатической системы: очень часто – нейтропения; часто – тромбоцитопения.
Нарушения со стороны печени и желчевыводящих путей: часто – повышение активности «печеночных» трансаминаз.
Нарушения со стороны обмена веществ и питания: часто гипертриглицеридемия, гиперхолестеринемия.
Общие расстройства и реакции в месте введения: часто – эритема и зуд в месте введения препарата.
Передозировка
Признаки и симптомы
Имеются ограниченные данные по передозировке препарат Кевзара®.
Лечение
Специфического лечения при передозировке препарата Кевзара® не существует. В случае передозировки следует тщательно контролировать состояние пациента, проводить симптоматическую и поддерживающую терапию.
Взаимодействие
Применение с другими препаратами для лечения ревматоидного артрита
Одновременное применение с метотрексатом не влияло на экспозицию сарилумаба. Не ожидается также влияния сарилумаба на экспозицию метотрексата при их одновременном применении, клинические данные отсутствуют.
Одновременное применение препарата Кевзара® с ингибиторами янус-киназы (ингибиторами JAK) или другими биологическими БМАРП, такими как антагонисты ФНО, антагонисты рецептора интерлейкина-1 (ИЛ-1R), анти-CD20 моноклональные антитела, селективные ко-стимулирующие модуляторы, не изучалось. Следует избегать одновременного применения препарата Кевзара® с биологическими БМАРП.
Взаимодействие с препаратами, являющимися субстратами цитохрома Р450
Различные исследования in vitro и ограниченное количество исследований in vivo на человеке показали, что цитокины и модуляторы цитокинов могут влиять на экспрессию и активность специфических изоферментов цитохрома Р450 (CYP) (CYP1A2, CYP2C19, CYP3A4) и, таким образом, имеют возможность изменять фармакокинетику одновременно принимаемых препаратов, являющихся субстратами для этих изоферментов. Повышение концентрации ИЛ-6 может снижать активность цитохрома Р450 у пациентов с ревматоидным артритом, и, следовательно, повышать у них концентрацию препаратов, являющихся субстратами цитохрома Р450, по сравнению с пациентами без ревматоидного артрита. Блокада сигнального пути ИЛ-6 антагонистами рецепторов ИЛ-6Rα, такими как сарилумаб, может устранить ингибирующее действие ИЛ-6 и восстановить активность цитохрома Р450, приводя к изменению концентрации лекарственных препаратов.
Изменение влияния ИЛ-6 на изоферменты цитохрома Р450 под действием сарилумаба может быть клинически значимо для субстратов цитохрома Р450 с узким терапевтическим диапазоном концентраций, для которых доза корректируется индивидуально. После начала применения или отмены препарата Кевзара® пациентам, получающим лечение лекарственными препаратами, являющимися субстратами цитохрома Р450, следует проводить мониторинг терапевтического эффекта (например, для варфарина) или концентрации лекарственного препарата (например, для теофиллина) и корректировать дозу лекарственного препарата по мере необходимости.
Следует соблюдать осторожность при начале терапии препаратом Кевзара® у пациентов, принимающих препараты, которые являются субстратами изофермента ЗА4 цитохрома Р450 (CYP3A4) (например, оральные контрацептивы или статины), так как сарилумаб может устранить ингибирующий эффект ИЛ-6 и восстанавливать активность изофермента CYP3A4, приводя к снижению экспозиции и активности субстратов CYP3A4. Взаимодействие сарилумаба с субстратами других изоферментов CYP (CYPP2C9, CYP2C19, CYP2D6) не изучалось.
Особые указания
Серьезные инфекции
В период лечения препаратом Кевзара® следует тщательно контролировать пациентов на предмет развития симптомов и признаков инфекций. Поскольку среди пациентов пожилого возраста частота развития инфекций выше, следует с осторожностью проводить лечение данной категории пациентов.
Препарат Кевзара® не следует применять у пациентов с активным течением инфекционного заболевания, включая локализованные инфекции. Необходимо оценить соотношение пользы и риска перед началом применения препарата Кевзара® у пациентов:
- с хронической или рецидивирующей инфекцией;
- с серьезными или оппортунистическими инфекциямив анамнезе;
- с ВИЧ-инфекцией;
- с сопутствующими заболеваниями, предрасполагающими к развитию инфекций;
- после контакта с больными туберкулезом;
- проживавших или посещавших регионы, эндемичные по туберкулезу или микозам.
Следует прервать лечение препаратом Кевзара®, если у пациента отмечается развитие серьезной или оппортунистической инфекции.
Пациент, у которого в период лечения препаратом Кевзара® развилась инфекция, должен незамедлительно пройти полное диагностическое обследование, предусмотренное для лиц с ослабленным иммунитетом; затем ему должна быть назначена адекватная антибактериальная терапия с последующим тщательным наблюдением.
У пациентов, получавших иммунодепрессивные препараты для лечения ревматоидного артрита, включая препарат Кевзара®, были зарегистрированы серьезные инфекции, иногда с летальным исходом, вызванные бактериальными, микобактериальными, инвазивными грибковыми, вирусными и другими оппортунистическими патогенами. Наиболее часто наблюдавшимися серьезными инфекциями при применении препарата Кевзара® были пневмония и целлюлит (воспаление подкожной жировой клетчатки). Из оппортунистических инфекций при применении препарата Кевзара® были зарегистрированы туберкулез, кандидоз и пневмоцистоз. В единичных случаях наблюдались диссеминированные, а не локализованные инфекции у пациентов, часто получающих сопутствующую терапию иммунодепрессивными препаратами, такими как метотрексат или глюкокортикостероиды, что в сочетании с ревматоидным артритом, может предрасполагать к развитию инфекции.
Туберкулез
До начала лечения препаратом Кевзара® у пациентов необходимо оценить наличие факторов риска туберкулеза и провести обследование на латентную инфекцию. Пациентам с латентным или активным туберкулезом следует до начала лечения препаратом Кевзара® провести стандартную противотуберкулезную терапию. У пациентов с латентным или активным туберкулезом в анамнезе, у которых невозможно подтвердить, проводился ли необходимый курс терапии, и у пациентов с отрицательным результатом анализа на латентный туберкулез, но имеющих факторы риска развития туберкулезной инфекции, следует рассмотреть возможность проведения противотуберкулезной терапии до начала лечения препаратом Кевзара®. При решении вопроса о проведении противотуберкулезной терапии целесообразно проконсультироваться с фтизиатром.
Следует тщательно контролировать пациентов на предмет развития признаков и симптомов туберкулеза, включая пациентов, чей результат обследования на латентный туберкулез до начала терапии был отрицательным.
Реактивация вирусной инфекции
При применении иммунодепрессивных биологических препаратов сообщалось о реактивации вирусных инфекций. В клинических исследованиях препарата Кевзара® отмечались случаи опоясывающего герпеса. В клинических исследованиях случаи реактивации вируса гепатита В зарегистрированы не были, однако из исследований были исключены пациенты, имеющие риск реактивации инфекции.
Лабораторные показатели
Количество нейтрофилов
При лечении препаратом Кевзара® отмечалась более высокая частота снижения АЧН. Снижение АЧН не сопровождалось более высокой частотой развития инфекций, включая серьезные инфекции.
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов с АЧН <2×109/л. У пациентов со снижением АЧН <0,5×109/л лечение препаратом Кевзара следует отменить.
Следует контролировать число нейтрофилов через 4-8 недель после начала терапии препаратом Кевзара® и далее – в зависимости от клинических показаний. Рекомендации по изменению дозы на основании значений АЧН представлены в разделе «Способ применения и дозы».
При изменении дозы препарата Кевзара® следует ориентироваться на показатели, полученные в конце интервала между введениями препарата.
Количество тромбоцитов
В клинических исследованиях при лечении препаратом Кевзара® отмечалось снижение количества тромбоцитов. Снижение количества тромбоцитов не сопровождалось развитием кровотечения.
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов с количеством тромбоцитов <150×103/мкл. При снижении количества тромбоцитов <50×103/мкл терапию препаратом Кевзара® следует отменить. Следует контролировать количество тромбоцитов через 4-8 недель после начала терапии и далее – в зависимости от клинических показаний. Рекомендации по изменению дозы препарата на основании количества тромбоцитов представлены в разделе «Способ применения и дозы».
Ферменты печени
При лечении препаратом Кевзара® отмечалась более высокая частота повышения активности «печеночных» ферментов, что в клинических исследованиях носило транзиторный характер и не приводило к появлению каких-либо клинически выраженных симптомов повреждения печени. Увеличение частоты и выраженности повышения активности «печеночных» ферментов наблюдалось при применении препарата Кевзара® в комбинации с потенциально гепатотоксичными препаратами (например, метотрексатом).
Не рекомендуется начинать лечение препаратом Кевзара® у пациентов с повышением активности «печеночных» трансаминаз аланинаминотрансферазы (АЛТ) или аспартатаминотрансферазы (ACT) >1,5×ВГН. При повышении активности АЛТ >5×ВГН терапию препаратом Кевзара® следует отменить.
Следует контролировать активность АЛТ и ACT через 4-8 недель после начала терапии и далее – каждые 3 месяца. В случае клинической необходимости следует рассмотреть возможность исследования других показателей функции печени, таких как билирубин. Рекомендации по изменению дозы на основании повышения активности «печеночных» трансаминаз представлены в разделе «Способ применения и дозы».
Изменение показателей липидного обмена
У пациентов с хроническими воспалительными заболеваниями концентрации липидов в крови могут быть снижены. Лечение препаратом Кевзара® сопровождалось повышением концентраций липидов, таких как холестерин ЛПНП, холестерин ЛПВП и/или триглицериды.
Необходимо контролировать показатели липидного обмена приблизительно через 4-8 недель после начала терапии препаратом Кевзара®, затем – примерно через каждые 6 месяцев.
Лечение пациентов осуществляется согласно клиническим руководствам по ведению пациентов с гиперлипидемией.
Перфорация желудочно-кишечного тракта
В клинических исследованиях было зарегистрировано такое нежелательное явление, как перфорация желудочно-кишечного тракта, которая, прежде всего, является осложнением дивертикулита. Следует с осторожностью применять препарат Кевзара® у пациентов, имеющих в анамнезе указания на язвы желудочно-кишечного тракта или дивертикулит. Необходимо незамедлительно обращать внимание на появление у пациентов новых абдоминальных симптомов, таких как стойкая боль и повышение температуры.
Злокачественные новообразования
Лечение иммунодепрессивными препаратами может приводить к увеличению риска развития злокачественных новообразований. Влияние терапии препаратом Кевзара® на развитие злокачественных новообразований неизвестно, но в клинических исследованиях были зарегистрированы случаи злокачественных новообразований.
Реакции гиперчувствительности
Сообщалось о развитии реакций гиперчувствительности, связанных с приемом препарата Кевзара®. Наиболее частыми реакциями гиперчувствительности были сыпь в месте введения препарата, кожная сыпь и крапивница. Пациента следует информировать о незамедлительном обращении к врачу в случае появления любых реакций гиперчувствительности. При развитии анафилактических реакций или реакций гиперчувствительности введение препарата Кевзара® следует прекратить немедленно. Препарат Кевзара® не назначают пациентам с известной гиперчувствительностью к сарилумабу.
Нарушение функции печени
Пациентам с активными заболеваниями печени или нарушениями функции печени лечение препаратом Кевзара® не рекомендуется.
Вакцинация
Следует избегать одновременного применения живых, а также живых аттенуированных вакцин во время лечения препаратом Кевзара®, поскольку клиническая безопасность данного взаимодействия не установлена. Отсутствуют данные о вторичной передаче возбудителей заболеваний от лиц, вакцинированных живыми вакцинами, пациентам, получающим препарат Кевзара®. Перед началом лечения препаратом Кевзара® рекомендуется, чтобы все пациенты были вакцинированы в соответствии с действующими рекомендациями по вакцинации. Интервал между вакцинацией живыми вакцинами и началом лечения препаратом Кевзара® должен соответствовать действующим рекомендациям по вакцинации в отношении одновременного применения иммунодепрессивных препаратов.
Кардиоваскулярный риск
Пациенты с ревматоидным артритом имеют повышенный риск развития сердечно-сосудистых заболеваний. Факторы риска (например, артериальная гипертензия, гиперлипидемия) необходимо учитывать в рамках проведения стандартной терапии.
Применение у детей
Не следует применять препарат Кевзара® у детей в возрасте до 18 лет (в настоящий момент безопасность и эффективность препарата не установлены).
Несовместимость
Ввиду отсутствия исследований на совместимость, препарат Кевзара® нельзя смешивать с другими лекарственными средствами.
Влияние на способность управлять транспортными средствами, механизмами
Препарат Кевзара® не оказывает или оказывает незначительное влияние на способность управлять транспортными средствами или работать с механизмами.
Условия хранения
При температуре от 2 °С до 8 °С в оригинальной упаковке для защиты от действия света. Не замораживать.
При извлечении из холодильника препарат должен храниться при температуре не выше 25 °С и быть использован в течение 14 дней.
Хранить в недоступном для детей месте.
Срок годности
2 года.
Препарат не следует применять по истечении срока годности.
Условия отпуска
Производитель
Санофи Винтроп Индустрия, Франция.
Организация, принимающая претензии потребителей
АО «Санофи Россия».
125009, г. Москва, ул. Тверская, 22.
Основные сведения
Торговое название
Кевзара
Действующее вещество (МНН)
Сарилумаб
Дозировка или размер
175 мг/мл
Форма выпуска
раствор для подкожного введения
Производитель
Sanofi-Winthrop
Условия хранения
В защищенном от света месте, при температуре 2–8 °C (не замораживать)
Представлено описание активных веществ лекарственного препарата. Описание препарата основано на официально утвержденной инструкции по применению от компании-производителя. Приведенное описание носит исключительно информационный характер и не может быть использовано для принятия решения о возможности применения конкретного лекарственного препарата.
Цены в аптеках на Кевзара 175 мг/мл, раствор для подкожного введения, 1,14 мл, 2 шт.

История стоимости Кевзара 175 мг/мл, раствор для подкожного введения, 1,14 мл, 2 шт.
04.09-10.09
64 441 ₽ (-523 ₽)
11.09-17.09
63 918 ₽ (-523 ₽)
18.09-24.09
64 964 ₽ (+1 046 ₽)
Указана средняя стоимость товара в аптеках региона Москва и МО за период и разница по сравнению с предыдущим периодом
Цены Кевзара и наличие в аптеках в Москве
175 мг/мл, раствор для подкожного введения, 1,14 мл, 2 шт.
| Список аптек | Адрес | Часы работы | Цена |
|---|---|---|---|
| 305 Аптека
ЗдравСити |
Сергиев Посад-6, 1А, Сергиево-Посадский р-н, МО (на въезде в Военный городок, на территории рынка) |
09:30-19:00 Пн-Пт, 09:30-17:00 Сб-Вс |
64 964 ₽ |
| 33 Аптека
ЗдравСити |
г.Сергиев Посад, площадь Красногорская, д. 1 |
10:00-19:00 Пн-Вс |
64 964 ₽ |
| 36,6
ЗдравСити |
Никольская ул, 10, Москва |
08:00-23:00 Пн-Вс |
64 964 ₽ |
| 468 Аптека
ЗдравСити |
г.Жуковский, Чкалова, д. 41 |
Круглосуточно |
64 964 ₽ |
| Apteka
ЗдравСити |
Курыжова, 7, Южный мкр, Домодедово, МО |
09:00-22:00 Пн-Вс |
64 964 ₽ |
| Ipharm
ЗдравСити |
Энтузиастов ш, 3 к 1, Москва |
Круглосуточно |
64 964 ₽ |
| REDApteka
Оплата только наличными ЗдравСити |
Семеновская пл, 7 к 17А, Москва |
09:00-21:00 Пн-Пт |
64 964 ₽ |
| А+А
ЗдравСити |
Юшуньская малая. ул, 3, Москва |
09:00-21:00 Пн-Вс |
64 964 ₽ |
| АBC
ЗдравСити |
Нахимовский пр, 22, Москва |
Круглосуточно |
64 964 ₽ |
| АВЕ
Оплата только наличными ЗдравСити |
Шелепихинская наб, 34 к 2, Москва |
10:00-22:00 Пн-Вс |
64 964 ₽ |
| Абикон Медика
ЗдравСити |
Армавирская ул, 3, Москва |
09:00-21:00 Пн-Сб, 10:00-21:00 Вс |
64 964 ₽ |
| Аве
Оплата только наличными ЗдравСити |
Пресненский Вал ул, 7c1, Москва |
10:00-22:00 Пн-Вс |
64 964 ₽ |
| Авилек
ЗдравСити |
Авиаконструктора Миля ул, 4А, Москва |
Круглосуточно |
64 964 ₽ |
| Авиценна
ЗдравСити |
Новоугличское ш, 46, Сергиево-Посадский р-н, Сергиев Посад, МО |
Круглосуточно |
64 964 ₽ |
| Авиценна Фарма
ЗдравСити |
Вучетича ул, 22, Москва |
Круглосуточно |
64 964 ₽ |
| Азбука Здоровья
Оплата только наличными ЗдравСити |
г.Москва, Михайлова, д. 8, корп. 1 |
09:00-22:00 Пн-Вс |
64 964 ₽ |
| № 17
ЗдравСити |
Рижское ш, 41А, Волоколамск, МО |
09:00-18:00 Пн-Пт, 09:00-15:00 Сб-Вс |
64 964 ₽ |
| № 458
ЗдравСити |
Ласточкин пр-д, 16, Балашиха, МО |
08:00-21:00 Пн-Вс |
64 964 ₽ |
| №1
ЗдравСити |
Маршала Жукова пр, 49, Москва |
09:00-21:00 Пн-Вс |
64 964 ₽ |
| №206 На Рязанском проспекте
ЗдравСити |
Рязанский пр, 71к1, Москва |
08:00-21:00 Пн-Пт, 09:00-19:00 Сб, 10:00-18:00 Вс |
64 964 ₽ |
Отзывы о Кевзара
У данного товара ещё нет отзывов
Ваш может стать первым!
Популярные товары в категории
Популярные товары в Ютеке
Купить Кевзара, 175 мг/мл, раствор для подкожного введения, 1,14 мл, 2 шт. в Москве с доставкой в аптеку или домой, сделав заказ через Ютеку
Цена Кевзара, 175 мг/мл, раствор для подкожного введения, 1,14 мл, 2 шт. в Москве от 64964 руб. на сайте и в приложении
Подробная инструкция по применению Кевзара, 175 мг/мл, раствор для подкожного введения, 1,14 мл, 2 шт.
Информация на сайте не является призывом или рекомендацией к самолечению и не заменяет консультацию специалиста (врача), которая обязательна перед назначением и/или применением любого лекарственного препарата.
Дистанционная торговля лекарственными препаратами осуществляется исключительно аптечными организациями, имеющими действующую лицензию на фармацевтическую деятельность, а также разрешение на дистанционную торговлю лекарственными препаратами. Дистанционная торговля рецептурными лекарственными препаратами, наркотическими и психотропными, а также спиртосодержащими лекарственными препаратами запрещена действующим законодательством РФ и не осуществляется.